ARTÍCULO ORIGINAL
RIBEIRO, Fernando de Sá [1], JESUS, Jéssica Barbosa de [2], SOUZA, Alessandra Mendonça Teles de [3]
RIBEIRO, Fernando de Sá. JESUS, Jéssica Barbosa de. SOUZA, Alessandra Mendonça Teles de. Análisis y caracterización de una prometedora diana terapéutica identificada en Leishmania spp. Revista Científica Multidisciplinar Núcleo do Conhecimento. Ano 05, Ed. 05, Vol. 09, pp. 99-132. Maio de 2020. ISSN: 2448-0959, Enlace de acceso: https://www.nucleodoconhecimento.com.br/salud/objetivo-terapeutico
RESUMEN
La leishmaniasis es una enfermedad desatendida causada por protozoos del género Leishmania spp., que afecta a alrededor de 1,6 millones de individuos cada año y 500.000 se presentan en la forma visceral. En Brasil hay alrededor de 30.000 nuevos casos cada año. Además, el país es responsable del 90% de los casos notificados de leishmaniasis visceral, y esta es la forma más grave de la enfermedad. Aliado a estos hechos, el tratamiento actual es ineficaz, contribuyendo al establecimiento de cepas resistentes. Actualmente, el tratamiento tiene varios efectos secundarios y daño permanente a la salud de los pacientes, este hecho ha contribuido a la búsqueda de nuevos fármacos contra la leishmaniasis. La enzima oligopeptidasa B (OPB) se ha estudiado como una posible diana terapéutica en el desarrollo de agentes antiparasitatorios. Así, el objetivo de este trabajo es construir el modelo tridimensional de la enzima Oligopeptidasse B de diferentes especies de Leishmania spp. y compararlos entre sí. Para ello, se utilizó el método de modelado comparativo. En este método, los modelos de la especie L. brasiliensis, L. donovani, L. infantum, L. mexicana y L. panamensis fueron construidos utilizando el programa MODELLER. Una vez que los modelos estaban listos, se llevó a cabo el proceso de validación y posteriormente se caracterizó, lo que fue posible verificar un grado prometedor de similitud entre los modelos. Finalmente, estos modelos fueron sometidos al método de análisis por modos normales, que obtuvieron un patrón de movimiento similar, por lo que fue posible verificar un movimiento en una región específica de una alfa-hélice, lo que llevó a la tríada de la enzima expuesta, lo que puede ser indicativo de un mecanismo de acción. Por último, se espera que utilice los modelos construidos para ayudar en el desarrollo de una nueva terapia prometedora para el tratamiento de la leishmaniasis.
Palabras clave: Leishmaniasis, oligopeptidasa B, modelado molecular, modos normales.
INTRODUCCIÓN
La leishmaniasis, causada por la leishmania spp., es una enfermedad caracterizada por varios tipos de manifestaciones, a partir de leves, en las que hay informes de lesiones pequeñas que incluso sin el debido tratamiento retroceden a las más graves como la Leishmaniasis Visceral (VL). En Brasil, la forma más grave de esta enfermedad presenta datos alarmantes en comparación con otros países, lo que convierte al país en el mayor titular de casos de VL en toda América (ALVARENGA, 2010; OMS, 2019).
Esta enfermedad pertenece al grupo de enfermedades desatendidas, que forman parte de todas aquellas enfermedades que afectan principalmente a los países subdesarrollados y a las regiones más pobres, por lo que no despierta interés en el desarrollo de medicamentos. Por lo tanto, es necesario que las técnicas eficientes y de bajo costo sobrevivan para superar esta falta de incentivos financieros para el estudio de esta enfermedad. Por lo tanto, los métodos computacionales se pueden utilizar con el fin de reducir el tiempo en el desarrollo de una nueva terapia prometedora y, en consecuencia, el costo en comparación con los métodos más tradicionales para el desarrollo de fármacos (BAILEY et al., 2017; OMS, 2017).
A pesar de la poca inversión en esta área, hay tratamiento para la enfermedad como el antimonio pentas valentes (fármaco de primera opción) o la anfotericina B (fármaco de segunda opción). Sin embargo, estos tratamientos tienen varias desventajas como la alta tasa de resistencia y la amplia variedad de efectos secundarios, que van desde el mareo hasta los posibles problemas causados al tercer (3er) par del nervio craneal, lo que conduce a dificultades motoras (MACEDO-SILVA et al., 2014).
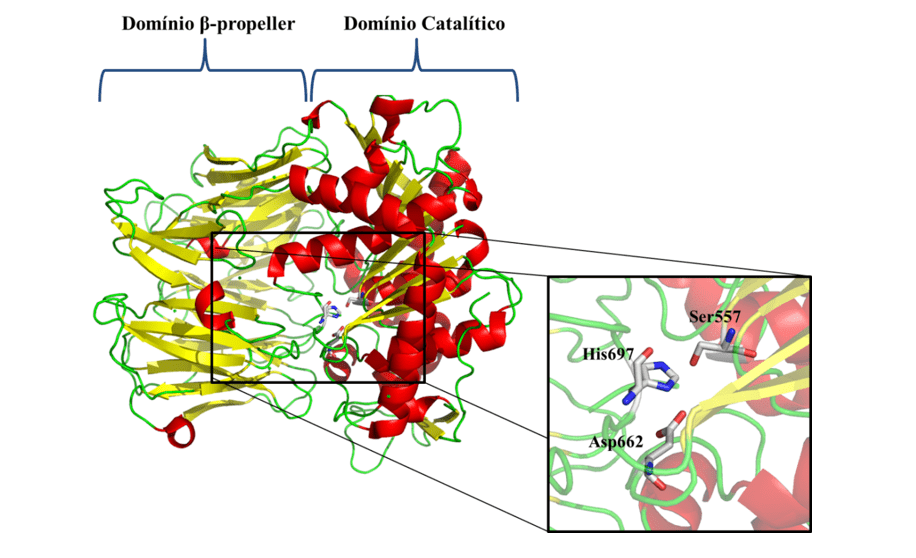
En vista de los problemas presentados y de la importancia epidemiológica de la leishmaniasis, no se escatoria la necesidad de investigar nuevas dianas terapéuticas específicas contra la enfermedad. Como una de las nuevas dianas terapéuticas, tenemos Oligopeptidasa B (OPB) (Figura 1), que es una proteasa de serina, perteneciente a la subfamilia S9A, teniendo como característica una tríada catalítica compuesta de los residuos de aminoácidos serina (Ser), ácido asáptico (Asp) e histidina (Su) que se encuentran entre los dos dominios. Esta enzima tiene como característica la capacidad de cortar residuos de estructuras proteicas. Además, se describe en la literatura como un componente clave para el mecanismo de escape inmune del parásito, cortando la enolase, una proteína que opsonisar el protozoo, de modo que cuando sea reconocido por el macrófago se destruye. Sin embargo, mientras que el parásito está dentro del macrófago, el OPB es súper expresado, haciendo que el parásito no sea reconocido dentro de la célula, por lo que se multiplicará hasta que se alisa (SODERO et al., 2016; SWENERTON et al., 2011; OVCHINNIKOVA et al. 2018).
Figura 1: Estructura tridimensional de L. principal OPB (código PDB 2XE4) (MCLUSKEY et al., 2010) que muestra los dominios catalítico y de hélice. Las estructuras secundarias, tales como la hélice, las hojas y las asas, se muestran en rojo, amarillo y verde, respectivamente. En particular, se muestran los residuos (Ser557, Asp662 e His697) de la tríada catalítica.
Por lo tanto, el estudio en cuestión tuvo como objetivo investigar y caracterizar los OPB y sus sitios activos, de la especie L. brasiliensis, L, donovani, L. infantum, L. mexicana y L. panamensis. Por último, se esperaba identificar posibles similitudes entre proteínas, de modo que permitiera el desarrollo futuro de una nueva terapia prometedora con un amplio espectro de acción sobre las enzimas de todas las especies en el estudio.
OBJETIVO GENERAL
En vista de la necesidad de desarrollar nuevas entidades químicas para el tratamiento contra la leishmaniasis, este trabajo tenía como objetivo principal construir y caracterizar la enzima oligopeptidasa B de Leishmania spp. utilizando técnicas de estudio computacional.
OBJETIVOS ESPECIFICOS
- Construir y validar los modelos de enzimas oligopeptidasa B de especies de Leishmania;
- Realizar la caracterización de enzimas;
- Realizar estudios de simulación por modos normales.
MATERIAL Y MÉTODOS
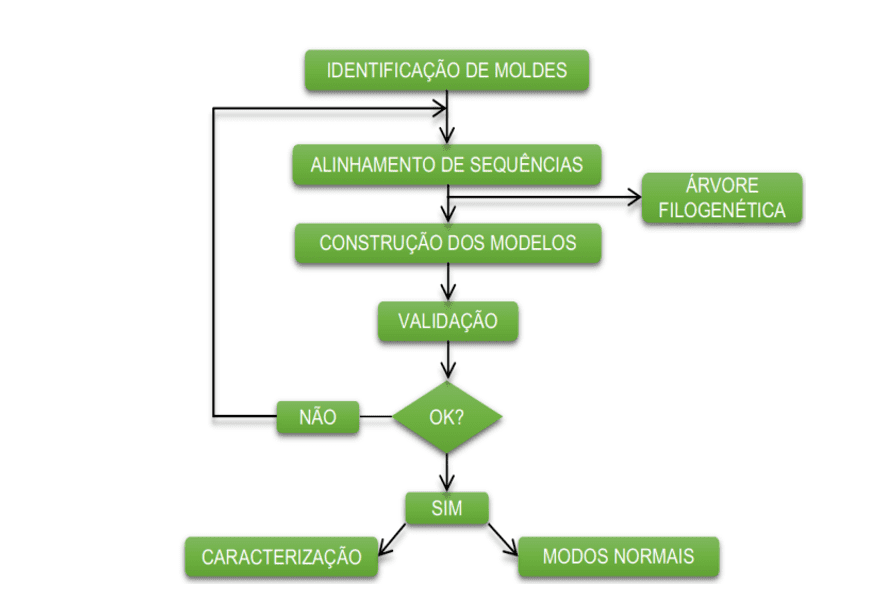
El diagrama siguiente (diagrama 1) muestra de forma simplificada los pasos utilizados durante el desarrollo de este trabajo.
Esquema 1: Esquema simplificado de pasos y métodos de material.
OBTENCIÓN DE LA ESTRUCTURA PRIMARIA
Las estructuras primarias de leishmanias OPBs se obtuvieron de la base de datos Uniprot (The Universal Protein Resource) (WANG et al., 2012).
ID DE MOLDE
Luego, la búsqueda de proteínas de moho se realizó a partir de la alineación entre la secuencia de aminoácidos de las secuencias de proteínas objetivo y aminoácidos de proteínas depositadas en el Banco de Datos de Proteínas (PDB) por el servidor BLAST (Basic Local Alignment Search Tool) (ATLSCHUL et al., 1997) basándose en la identidad entre secuencias de aminoácidos.
ALINEACIÓN DE SECUENCIAS
Una vez definido el molde a utilizar, la secuencia de moldes se alineó con sus respectivos OPB, en el programa ClustalOmega (ClustaO) (SIEVERS et al., 2011).
CONSTRUCCIÓN DE MODELOS
Después de obtener la alineación, esta información se utilizó en el programa MODELLER v9.20 para construir las estructuras 3D de los modelos (WEBB y SALI, 2016).
VALIDACIÓN DE MODELOS CONSTRUIDOS
Para la validación de los modelos, se utilizó el gráfico Ramachandran generado por el servidor PDBsum, Verify 3D y ProSA-web, todo obtenido del servidor Saves.
En el análisis del gráfico Ramachandran, es permisible visualizar los ángulos diédricos (phi (o) y psi (o)) de los residuos de aminoácidos en la estructura proteica. El ángulo phi (o) es el resultado del vínculo entre el grupo NH y el carbono alfa, mientras que el ángulo psi (o) se origina en el vínculo entre el carbono alfa y el grupo carbonilo (RAMACHANDRAN et al., 1963).
El gráfico proporciona una manera fácil de visualizar la distribución de los ángulos de larm de una estructura proteica. Además, proporciona una visión general de las regiones permitidas y no permitidas de los valores de ángulo de torción, sirviendo como un factor importante en la evaluación de la calidad de las estructuras de proteínas 3D. Con esto, define los residuos que se encuentran en las regiones que son energéticamente más favorables y desfavorables y guía la evaluación de la calidad de los modelos teóricos o experimentales de proteínas. Este gráfico se divide en regiones, de modo que las regiones más favorables están en rojo, regiones adicionales más permisivas en regiones marrones, permisivas en regiones amarillas y no permisivas en blanco. Según esta validación para un modelo pronosticado para ser considerado de excelente calidad, debe tener más del 90% de residuos de aminoácidos en la región más favorable (SANTOS-FILHO y ALENCASTRO, 2003).
Además, necesita tener la mayoría de sus residuos en las regiones más favorables, así como no tener residuos en las regiones no permisivas, excepto los aminoácidos glicina (Gly) y prolina (Pro) que son excepciones para esta área. Estos dos residuos presentan variaciones en la cadena lateral que confieren una mayor rigidez, en el caso de Pro, y una mayor flexibilidad, en el caso de Gly, siendo así puede asumir angulaciones inesperadas. Por esta razón se aceptan en las regiones no permisivas del gráfico. La existencia de las regiones no permitidas se debe al hecho de que hay efectos estetéricos entre los residuos (cadenas laterales) de aminoácidos. (MORRIS et al., 1992).
Verificar 3D analiza la compatibilidad de un modelo atómico (3D) con su propia secuencia de aminoácidos (1D). Cada residuo recibe una clase estructural basada en su ubicación y entorno (alfa, beta, bucle, polar, apolar, etc.). Poco después, una base de datos generada a partir de buenas estructuras se utiliza para obtener una puntuación para cada uno de los 20 aminoácidos en esta clase estructural. El eje vertical en el gráfico representa la puntuación media del perfil 3D-1D para cada residual en una ventana deslizante de 21 residuos y los resultados en forma de puntuaciones oscilan entre -1 (puntuación pobre) a +1 (buena puntuación) (EISENBERG et al., 1997).
ProSa-web calcula una puntuación de calidad general para una estructura de entrada específica. Si la puntuación de esto está fuera de un rango característico de las proteínas nativas, la estructura probablemente contiene errores. Una gran cantidad de puntuaciones de calidad local apuntan a partes problemáticas del modelo que también se resaltan en un visor de moléculas 3D para una fácil detección (WIEDERSTEIN y SIPPL, 2007).
ANÁLISIS DE LA ESTRUCTURA SECUNDARIA
Después de la etapa de validación, las estructuras terciarias de los modelos se compararon con la distribución de la estructura secundaria predicha por Quick2D, disponible en el servidor Bioinformatics Toolkit (https://toolkit.tuebingen.mpg.de/), eligiendo así los modelos (ALVA et al., 2016).
CARACTERIZACIÓN DE OLIGOPEPTIDASES (OPBS) DE LEISHMANIAS
MAPA POTENCIAL ELECTROSTÁTICO (MEP)
Para obtener los eurodiputados de las superficies y los sitios de conexión de los OPB, se utilizó una extensión del programa PyMOL, herramientas apb (BAKER et al., 2001). Antes de ser analizadas en PyMOL, las enzimas se prepararon en el servidor PDB2PQR (http://nbcr-222.ucsd.edu/pdb2pqr_2.0.0/) utilizando el parámetro de servidor estándar (DOLINSKY et al., 2004).
CARACTERIZACIÓN DE SITIOS Y SUBSITIOS DE OPBS
Los modelos y el molde fueron presentados en la plataforma de proteínas más utilizando la opción Dogsitescorer (VOLKAMER et al., 2012), en la que se predijeron cavidades en las estructuras 3D de los modelos. Esto generó resultados con respecto a posibles sitios de unión y subsitios enzimáticos para la predicción de estas cavidades. El programa hace uso de una malla tridimensional cuyo borde se puede ajustar entre 0,2o y 1,0o, junto con un filtro gaussiano que se utiliza para identificar cavidades en la superficie de la proteína que son adecuadas para acomodar átomos ligando. Además, DoGSiteScorer también predice la capacidad de farmacológica para cada cavidad pronosticada. Por lo tanto, para cada interacción entre la proteína y el fármaco posible, se asigna una puntuación que se refiere a la farmacabilidad de la cavidad, llamada drugscore. Para predecir el valor de drugscore, el programa utiliza una máquina de vectores de soporte (SVM), en la que se utilizan los siguientes descriptores: volumen, proporción de residuos no polares y profundidad (VOLKAMER et al., 2012).
TREE FILOGENÉTICO
Finalmente, el grado de parentesco evolutivo entre especies de Leishmanias se analizó utilizando el programa MEGA (Molecular evulutionary genetics analysis), utilizando los métodos Neighbor-joining, que permite la construcción del árbol filogenético con el fin de definir las ventajas evolutivas entre las poblaciones de secuencias previamente definidas por el usuario (KUMAR et al., 2004).
MODOS NORMALES
Para realizar los modos normales, se utilizaron los archivos generados en las fases de minimización de energía realizadas por GROMACs versión 5.1.2. Se realizaron los primeros 4 pasos relacionados con el proceso de dinámica molecular. La primera fue la generación de archivos de topología, con la adición de hidrógenos a la proteína. En el segundo, se creó la caja de agua, y este es un paso muy importante para el cálculo de la interacción entre la proteína y el disolvente. En el tercero, se añadieron iones para establecer un sistema neutral. Finalmente, en la cuarta etapa, se realizaron minimizaciones de energía, en las que se insertó el campo de fuerza AMBER99SB. A partir de este punto, los modos normales de los modelos se realizaron utilizando el servidor ANM (Anisotropic Network Model) (http://anm.csb.pitt.edu/) con el fin de analizar el movimiento de enzimas de las especies de Leishmania y también para observar algunas características importantes para las enzimas, tales como posibles movimientos relacionados con el mecanismo de acción (EYAL et al., 2015).
RESULTADOS Y DISCUSSIONS
SELECCIÓN DE PROTEÍNAS DE MOHO Y ALINEACIÓN ENTRE SECUENCIAS
Obtuvimos 100 estructuras primarias de los OPB de leishmania spp. utilizando el servidor UniProt, con 5 especies de Leishmanias seleccionadas. Las secuencias de aminoácidos revisadas seleccionadas de la enzima OPB fueron L.brasiliensis, L. donovani, L. infantum, L. mexicana y L. panamensis bajo los códigos A4H5Q8, C9EF60, A4HTZ8, E9AMS8 y A0A088RJA7, respectivamente Estas especies fueron seleccionadas, debido a su alta incidencia en América del Sur y su resistencia contra el tratamiento actual para la leishmaniasis (GHORBANI y FARHOUDI, 2018).
Para la construcción de modelos 3D, el programa BLAST se utilizó para comparar las secuencias de aminoácidos de las secuencias objetivo con secuencias de proteínas de estructuras tridimensionales aclaradas experimentalmente. Basándose en la identidad entre las secuencias y el número de huecos, el ENZYME OPB de L. major (código PDB 2XE4) fue seleccionado como proteína de molde (MCLUSKEY et al., 2010). La identidad entre las enzimas diana y sus respectivos moldes presentaba valores entre el 86% y el 96% (Tabla 1). El porcentaje de identidad entre dos secuencias se refiere a la presencia del mismo aminoácido en la misma posición entre las secuencias alineadas. Para la construcción de un modelo de una proteína con más de 80 residuos de aminoácidos, el porcentaje de identidad entre las estructuras primarias del molde y el modelo debe ser superior al 25%. Además, el porcentaje de brechas debe ser bajo del 20% para ser considerado una buena alineación (SANTOS-FILHO y ALENCASTRO, 2003). Por lo tanto, la probabilidad de similitud de las estructuras tridimensionales de las proteínas es alta.
Tabla 1: Porcentaje de identidad entre los modelos de oligopeptity B de las especies de Leishmania y su respectivo moho.
| Proteína molde (código PDB) | Modelos de OPB
(código uniprot) |
Identidade (%) | Gaps (%) |
| OPB
L. major (código PDB 2XE4) |
L.brasiliensis
(A4H5Q8) |
86 | 0 |
| L. donovani
(C9EF60) |
96 | 0 | |
| L. infantum
(A4HTZ8) |
96 | 0 | |
| L. Mexicana
(E9AMS8) |
90 | 0 | |
| L. panamensis
(A0A088RJA7) |
86 | 0 |
Fuente: Autor.
En el análisis de árboles filogenéticos, fue posible explicar el grado de identidad entre las especies de leishmania. Se observó que las especies con mayor grado de identidad, como L. infantum y L. donovani, ambas con 96%, presentaban una proximidad entre sí, además de estar más cerca de L.major. Las otras especies presentaban valores más bajos como L.mexicana (90%), que estaba en la mediana en relación con los otros modelos. Finalmente, los modelos con el menor porcentaje de identidad, L. brasiliensis y L. panamensis, ambos con 86%, estaban más distantes evolutivamente de su moho, pero estaban muy cerca el uno del otro (Figura 2). Este análisis permitió comprender la diferencia entre el grado de identidad entre las especies y la comprensión de algunas características importantes de la enzima entre la especie.
Figura 2: Esquema de árbol filogenético de las enzimas Leishmania spp. En esta representación, se evidencian los grupos con la mayor similitud entre sí, donde el grupo 1 se resalta en rojo y el grupo 2 en verde.
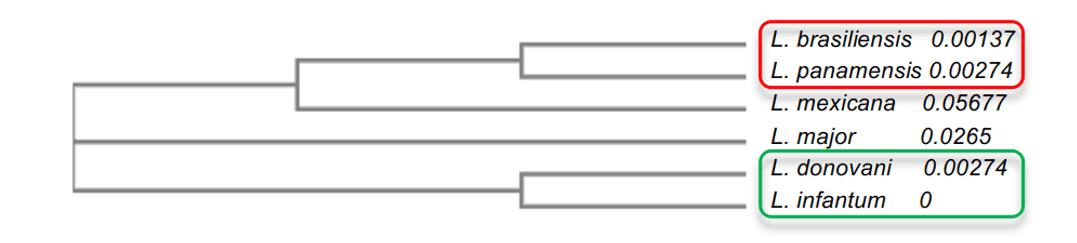
MODELOS TRIDIMENSIONALES DE LA ENZIMA DE LAS ESPECIES DE LEISHMANIA
Los modelos 3D de la especie fueron construidos utilizando el programa Modeller (WEBB y SALI, 2016) a partir de la alineación de las estructuras primarias (figura 3 y 4).
Figura 3: Resultado de la alineación entre las secuencias primarias de los OPB seleccionados en UNIPROT (Parte 1/2).

Figura 4: Resultado de la alineación entre las secuencias primarias de los OPB seleccionados en UNIPROT (Parte 2/2).

Al observar el resultado de la alineación de las estructuras (Figuras 3 y 4), se observó un mayor grado de similitud entre L. donovani y L. infantum (grupo 2), así como una mayor correspondencia entre L. brasiliensis y L. panamensis (grupo 1). Estos resultados refuerzan lo que fue presentado por el árbol filogenético en la Figura 2, donde se sugiere una proximidad entre las especies, L. donovani y L. infantum y otra proximidad entre las especies de L. brasiliensis y L. panamensis. Por otro lado, los dos grupos mencionados presentaban una mayor distancia evolutiva en comparación entre sí. Este hecho es notorio cuando se hace una comparación en el análisis de alineación, donde las estructuras primarias mostraron una mayor diferencia entre los residuos, en comparación con los dos grupos. Además, el resultado de la alineación de L. mexicana resultó ser muy prometedor, lo que se refiere al apoyo de este debate propuesto, ya que las diferencias observadas en comparación con las otras especies eran equivalentes, a veces al primer grupo mencionado, a veces al segundo. En algunas partes de la secuencia analizada, también se observaron mutaciones específicas que sólo son características de L. mexicana en algunas partes de la secuencia analizada. Por lo tanto, este hecho puede ser atestiguado debido a su posición en la mediana en el árbol filogenético, en relación con las otras especies.
Como resultado del modelado comparativo, se obtuvieron los modelos del presente estudio, lo que sugirió una gran similitud visual en su forma tridimensional en relación con el molde (Figura 5). En cuanto a la comparación entre los modelos, presentaron diferentes cantidades en las estructuras de hélice y hoja entre ellos. Esta diferencia puede explicarse por las diferencias específicas en la composición de los residuos de proteínas, pero tales diferencias no se presentaron en regiones importantes de estas enzimas, como el sitio de unión. Por lo tanto, estas diferencias no son tan importantes como para modificar las estructuras o el perfil de interacción con un posible medicamento en el sitio de enlace.
Figura 5: Modelos y molde de los OPB de leishmania spp., siendo evidenciado en rojo las hélices, verdes las asas y amarillo las hojas- . Para OPB, (A) L.major (molde) e modelos: (B) L. brasiliensis, (C) L. donovani, (D) L. infantum, (E) L. mexicana e (F) L. panamensis.
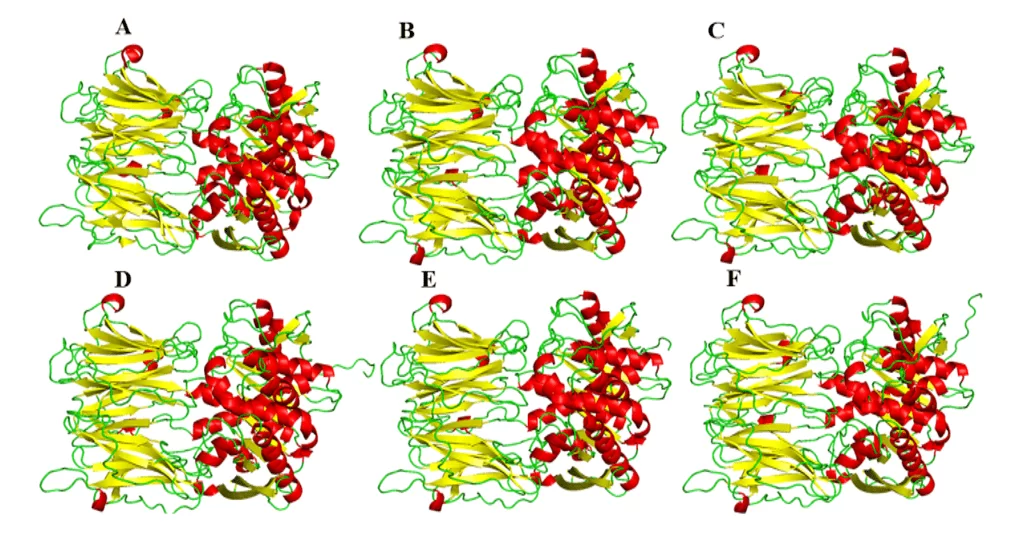
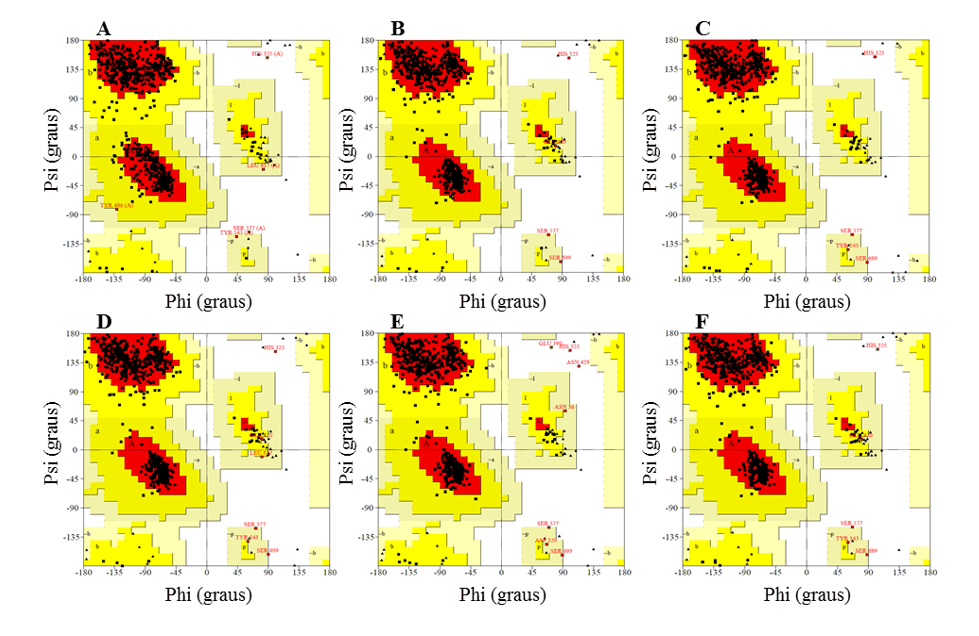
En la etapa de validación, se analizaron basándose en el análisis gráfico ramachandran (proporcionado por el servidor PDBsum), la puntuación 3D-1D (proporcionada por el programa verify-3D) y la puntuación Z (proporcionada por el servidor web ProSA). Los valores obtenidos para los modelos se compararon con los obtenidos para el molde.
En el gráfico Ramachandran, los modelos presentaban la mayoría de residuos en las regiones favorables, que oscilaban entre el 91,2 y el 92,3%, mientras que el porcentaje de residuos en regiones desfavorables era de un máximo del 0,5%, y los mejores modelos eran L.infantum y L.ienses. Estos modelos presentaron el mayor número de residuos en regiones favorables con 92,3% y 92,2% y el menor porcentaje de residuos en regiones desfavorables con 0,3% y 0,2%, respectivamente (Figura 6).
En todos los modelos fue posible observar que el residuo Ser, que forma parte de la tríada catalítica, se encontraba en las regiones desfavorables. Sin embargo, este hecho no afecta a la validez de los modelos, ya que al comparar el molde presentó el mismo resultado. Por lo tanto, este resultado no configura una confiabilidad baja de los modelos.
Figura 6: Resultados de los gráficos ramachandran, obtenidos por el programa PROCHECK, las estructuras de los modelos DE OPB generados y el molde.
| Estructuras | % de residuos en regiones | ||||
| Favorable | Permitido | Desfavorable | |||
| OLIGOPEPTIDASE B | L. major (A)
(PDB 2XE4) |
90,2 | 9,5 | 0,3 | |
| L.brasilienses (B) | 92,2 | 7,7 | 0,2 | ||
| L. donovani (C) | 91,9 | 8,0 | 0,2 | ||
| L. infantum (D) | 92,3 | 7,3 | 0,3 | ||
| L. Mexicana (E) | 91,2 | 8,3 | 0,5 | ||
| L. panamensis (F) | 91,4 | 8,3 | 0,3 | ||
Fuente: Preparado por el autor en base a los resultados de procheck.
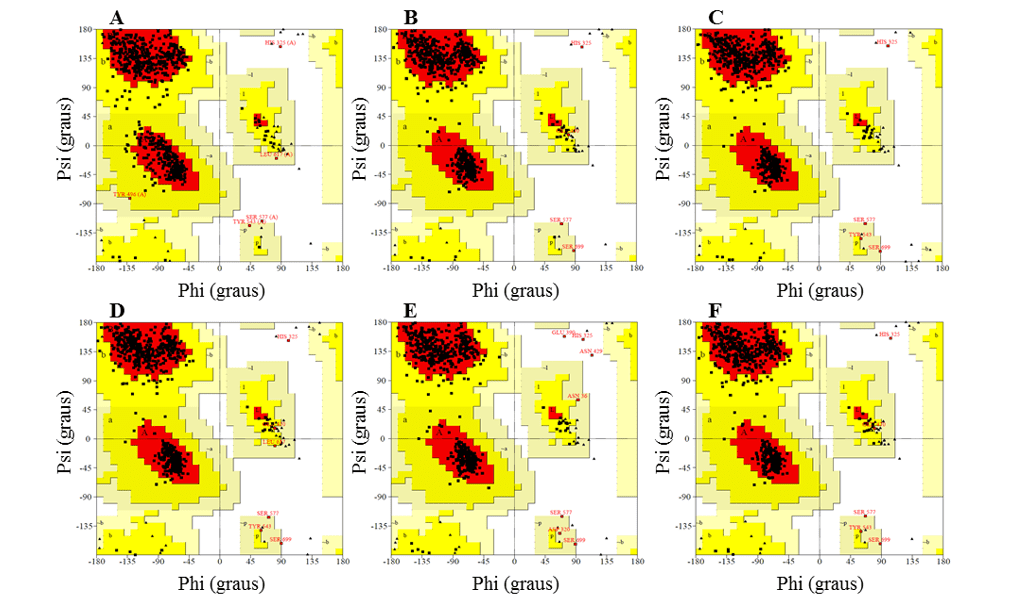
Cuando se utiliza el servidor Web ProSA de los modelos generados por MODELLER, presentaron valores de score-Z -10.84 a -11.19 y estos valores son compatibles con las estructuras PDB (Figura 7).
Figura 7: Resultados de la puntuación Z calculados en el servidor Web ProSA de las estructuras de molde (para la comparación). (A) L .major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. La región en azul oscuro indica la puntuación de las proteínas obtenidas por RMN y en azul claro de las proteínas obtenidas por difracción de rayos X.
El programa Verificar 3D se utilizó para evaluar la compatibilidad entre estructuras 1D y 3D. Los modelos obtenidos presentaron entre el 95,28% y el 98,34% de los aminoácidos con compatibilidad 3D-1D> 0,2, y los modelos L. donovani y L.brasilienses obtuvieron mejores resultados. De acuerdo con los parámetros ideales del programa, la mayoría de los residuos deben presentar valores por encima de cero, ya que los valores por debajo de cero indican regiones de la molécula con problemas. El porcentaje de aminoácidos con compatibilidad 3D-1D> 0,2 debe estar por encima del 80% (EISENBERG et al., 1997). Por lo tanto, estos resultados indican que los modelos presentaron compatibilidad 1D-3D, y los residuos que presentaban incompatibilidad no forman parte del sitio activo de las enzimas (Tabla 2).
Tabla 2: Resultados de Verificar 3D, mostrando el porcentaje de residuos con puntuación > 0.2.
| Estruturas | % de resíduos com score > 0,2 | |
| OLIGOPEPTIDASE B | L.major
(PDB 2XE4) |
93,20 |
| L.brasilienses | 95,62 | |
| L.donovani | 97,12 | |
| L.infantum | 94,93 | |
| L.mexicana | 95,62 | |
| L.panamensis | 94,93 |
Fuente: Autor.
VALIDACIÓN DE LA TRÍADA CATALÍTICA DE LA ENZIMA DE LOS MODELOS
Debido al mecanismo de PB, es esencial analizar la distancia y orientación entre los residuos de aminoácidos y la tríada catalítica (Ser, Asp y la suya) de los modelos generados con el fin de aumentar la fiabilidad de los modelos.
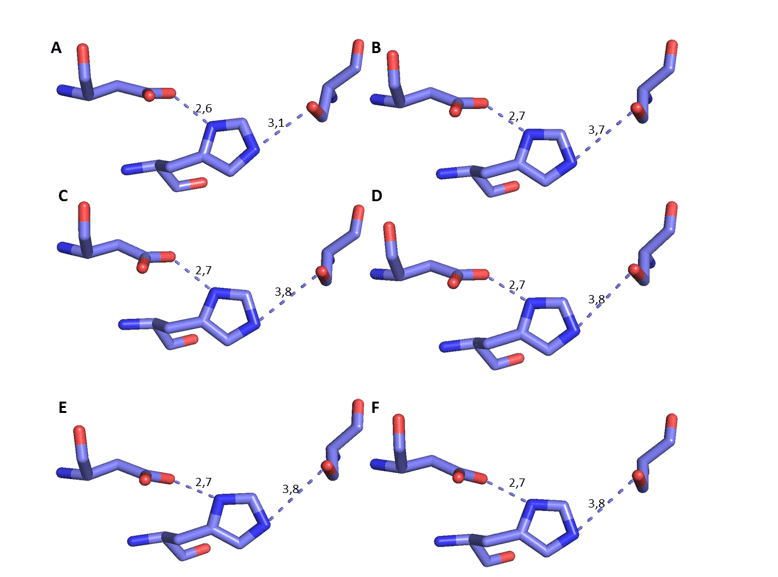
Es posible observar la comparación de estos residuos específicos de los modelos OPB con el molde en cuestión en la figura 17. Además, fue posible medir la distancia entre los residuos de la tríada catalítica, basándose en el mecanismo de interacción entre el sitio y el sustrato, con el suyo como referencia (DEREWENDA et al. , 1994). Por lo tanto, debe haber una distancia específica al mecanismo de acción. Esta distancia debe ser de aproximadamente 3,5o entre su oxígeno y el nitrógeno Ser, además de la distancia de aproximadamente 2,6o entre el oxígeno aspartato y su otro nitrógeno, como se describe en la literatura (DEREWENDA et al. , 1994). En este análisis, fue posible verificar que la distancia en cuestión se sometió a una pequeña variación, y todavía está de acuerdo con lo que ya se ha descrito. Esta distancia es importante porque las proteasas de serino requieren su escote de sustrato (HEDSTROM, 2002) (Figura 8). Sobre la base de estos resultados fiables, se continuó la caracterización de los modelos.
Figura 8: Representación 3D de la tríada catalítica de LOS OPB, respectivamente, Ser, Su y Asp. El color azul oscuro representa el átomo de nitrógeno, el oxígeno rojo y el carbono lila. Para OPB, (A) L.major (molde)e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis.
CARACTERIZACIÓN DE OLIGOPEPTIDASES DE LEISHMANIAS
ESTRUCTURA SECUNDARIA DE PREDICCIÓN
A partir de la predicción de la estructura secundaria de las enzimas OPB, se reveló el número y la posición de las estructuras secundarias de los modelos. El PSISPRED presentó entre 15 y 16 estructuras de hélice y todas presentaron 38 pares de hojas de é (Tabla 3). A partir de los resultados generados por PSIPRED, se realizó una comparación visual de esta estructura secundaria pronosticada con las estructuras tridimensionales de los 5 modelos, utilizando el programa Pymol. Los modelos de L.brasilienses, y L. panamensis presentaron la mejor similitud, en relación con las estructuras predesticias secundarias y terciarias (2D-3D), presentando 15 hélices y 38 hojas. De las 15 hélices presentadas en la estructura tridimensional del modelo 8 de las hélices de la hélice estaban en la misma posición que predijo el PSIPRED. Estas hélices corresponden a la secuencia de aminoácidos: 59 a 74, 78 a 93, 535 a 540, 545 a 563, 631 a 640, 703 a 721 y 727 a 730.
En el análisis general, no hubo diferencias exacerbadas en las cantidades de las hojas de hélice y de hélice entre las predicciones y los modelos obtenidos (Tabla 3).
Tabla 3: Comparación entre las estructuras secundarias predichas por psipred y las encontradas a través de Pymol.
| Modelos | α-Hélices | Folhas β | PSIPRED
α-Hélices |
PSIPRED
Folhas β |
| L.brasiliensis | 15 | 38 | 11 | 36 |
| L. donovani | 16 | 38 | 11 | 36 |
| L. infantum | 16 | 38 | 11 | 36 |
| L. mexicana | 15 | 38 | 10 | 36 |
| L. panamensis | 15 | 38 | 11 | 36 |
Fuente: Autor.
Por otra parte, fue posible observar que la cantidad de 15 hélices era la misma para L.brasilienses, L. Mexicana y L. panamensis. Al igual que L.infantum y L.donovani, tienen el número 16 de Helix. Según el árbol filogenético, estos dos grupos mencionados están en el mismo nodo interno y se consideran monofiléticos (Figura 2).
A continuación, se realizó el RMSD (desviación media-cuadrada de raíz) entre los modelos y el molde. Los valores eran prometedores, como se puede ver en el Cuadro 4, porque los RMSD no excedían el valor de 0,19o. Este hallazgo puede justificarse por el alto grado de identidad entre el molde y los modelos respectivos. En general, se espera que las proteínas con una identidad superior al 30%, tengan una excelente superposición de las cadenas principales, obteniendo así un RMSD del orden de 2o (BENNER et al., 1997; CHOTHIA et al., 1986).
Tabla 4: RMSD de los OPB generados por el Modelador, teniendo con orientación los carbonos alfa del molde OPB de L. mayor.
| Molde | Modelos | RMSD (Å) |
| L. major
(2XE4) |
L.brasiliensis | 0,15 |
| L. donovani | 0,15 | |
| L. infantum | 0,16 | |
| L. Mexicana | 0,19 | |
| L. panamensis | 0,14 |
Fuente: Autor.
MAPA DEL POTENCIAL ELECTROSTÁTICO MOLECULAR (MEP) DE LA SUPERFICIE DE ENZIMAS Y SITIOS RECEPTIVOS
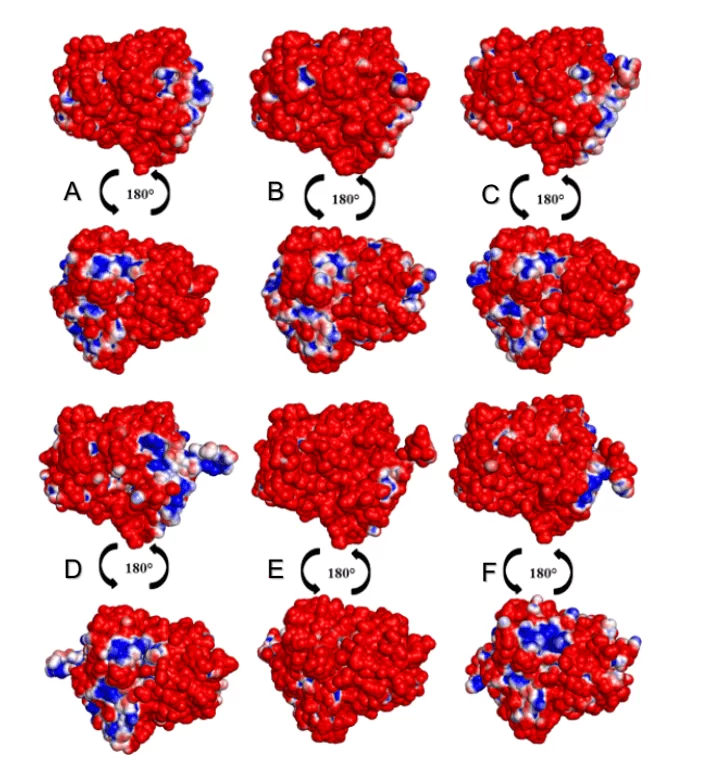
En el análisis de los eurodiputados de las superficies de los OPB, fue posible observar que todas las especies de leishmania presentaban un mayor porcentaje de regiones negativas que positivas, como se muestra en la Figura 15. Las especies L. donovani y L. infantum (grupo verde) sugirieron un área negativa (en color azul) en la misma región. Las especies L.brasilienses y L. panamensis (grupo rojo) presentaron una región negativa similar, en un patrón colorimétrico similar. Ambos resultados pueden justificarse por el hecho de que las especies comparadas entre sí pertenecen al mismo monofilético (Figura 2).
Figura 9: Mapa del potencial electrostático de los modelos 3D de Leishmania spp OPBs y el de su molde. Para OPB, (A) L.major (molde) e modelos: (B) L. brasilienses (C) L. donovani, (D) L. infantum, (E) L. mexicana (F) L. panamensis. En el color azul presenta la región positiva y en el color rojo, la región negativa.
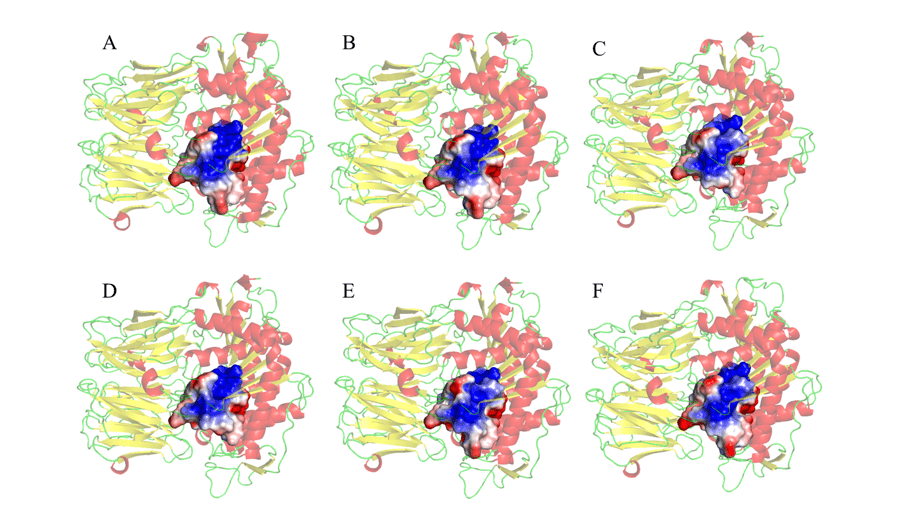
Por último, el mep de los residuos también se realizó en un radio de 5o alrededor de la tríada catalítica, como se muestra en la Figura 10. La elección de observar el MEP de la región alrededor de la tríada catalítica fue porque esta es la segunda región de interacción entre la enzima y el sustrato, en la que habrá alojamiento (ANDERSSON et al., 2010). Así, en vista de uno de los objetivos del estudio, fue posible verificar similitudes y correspondencias significativas entre el molde y los modelos probados. Estos resultados, como se muestra en la Figura 10, revelaron principalmente una porción electropositiva más grande (en azul) en la región central de los sitios de conexión. Se observaron regiones negativas (en rojo) en las zonas periféricas de los eurodiputados estudiadas. Estos resultados son prometedores, ya que se encontró que estas regiones son totalmente similares en todos los modelos, y pueden ayudar en el desarrollo de un medicamento que tiene la capacidad de actuar específicamente en todos los modelos estudiados.
Figura 10: Representación del mapa potencial de la superficie electrostática de residuos de aminoácidos, 5o alrededor de la tríada catalítica que componen el sitio activo de la enzima. (A) L.major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. En rojo son las hélices, de color verde las asas y de color amarillo las hojas. En el color dorado el sitio de encuadernación. En el color azul presenta la región positiva y en el color rojo, la región negativa.
DOGSITESCORER
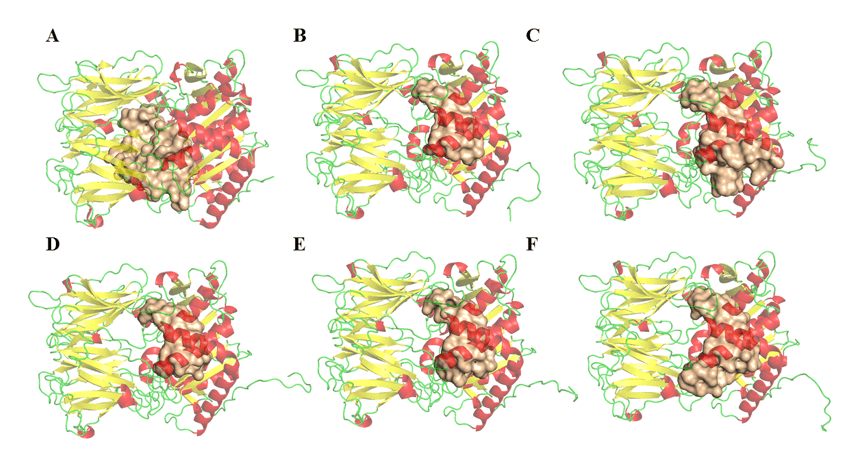
La determinación de los parámetros de volumen, área y profundidad de los posibles sitios de unión de las enzimas opb de las especies de Leishmania se realizó en el programa DoGSiteScorer (http://poseview.zbh.uni-hamburg.de/) de ProteinsPlus – Structure-Based Modeling Support Server (VOLKAMER et al., 2012). El programa señala tres sitios de enlace como se describe en la literatura. La figura 11 muestra la comparación entre la región posible del sitio activo y sus diferencias. Estas variables entre los OPOP pueden estar relacionadas con residuos que no se describen como importantes para su inhibición, pero se consideraron durante este análisis. Por lo tanto, este resultado no descarta la posibilidad de antipaína o cualquier otra molécula que tenga un amplio espectro de inhibición en la enzima, debido al hecho de que los resultados relacionados con la puntuación de fármacos fueron muy similares y positivos en todas las regiones. Se observa la Tabla 5, una discrepancia relativa entre el volumen y los resultados de área de las cavidades encontradas en los modelos. Este resultado no sugiere un sesgo de duda sobre el potencial inhibitorio de un solo medicamento en las enzimas respectivas. Sin embargo, presta atención al hecho de que algunas proteínas tienen cavidades más grandes que otras. Sin embargo, estas mismas cavidades tienen puntos en común, y estos son los residuos que componen el sitio catalítico, como se puede ver en la Figura 11 y se pueden explorar.
Tabla 5: Valores que hacen referencia a las posibles regiones de conexión de los OPB (obtenidos por el servidor).
| Estruturas | DogSiteScoore | |||
| Volume | Área | Drug Score | ||
| OLIGOPEPTIDASE B | L. major
(PDB 2XE4) |
1690,62 | 1818,41 | 0,80 |
| L.brasiliensis | 1527,84 | 1766,63 | 0,80 | |
| L. donovani | 1074,57 | 1428,55 | 0,79 | |
| L. infantum | 1309,97 | 1572,19 | 0,80 | |
| L. mexicana | 800,92 | 799,38 | 0,85 | |
| L. panamensis | 971,96 | 1083,40 | 0,81 | |
Fuente: Autor.
Figura 11: Estructuras de OPB y posibles regiones de conexión (obtenidas por el servidor DogSite). (A) L .major (molde)e modelos: (B) L. brasilienses, (C), L. donovani, (D) L. infantum, (E) L. mexicana e (F) L. panamensis. En rojo son las hélices, de color verde las asas y de color amarillo las hojas. En el color dorado el sitio de encuadernación.
MODOS NORMALES
Después de la caracterización de la enzima en los sitios estructurales, superficiales y de unión, los modos normales para las enzimas de cada especie se realizaron con el propósito de sus respectivos movimientos.
Después de la relajación y la minimización de la energía realizadas por la dinámica molecular en GROMACS, estas estructuras se sometieron a análisis normales con el fin de observar posibles movimientos compatibles.
Fue posible observar en todos los modelos estudiados un movimiento expresivo de una hélice específica, y al analizar esta región en su composición de aminoácidos es posible notar que está altamente conservada. Por lo tanto, el patrón de movimiento se repitió en todos los modelos en cuestión. Esta región sugirió un movimiento lineal que se alejaba del centro hacia la periferia, exponiendo la tríada catalítica. Esto puede ser indicativo del movimiento realizado por la proteína para el alojamiento del sustrato.
Al observar las figuras 12 y 13, se puede observar el mismo patrón de color en todos los modelos del estudio. Estos datos representan la capacidad que tiene la región dada para moverse en una dirección determinada. Los colores azules representan regiones más invariables durante la simulación por modos normales, con esto, un patrón de rigidez en las proteínas es notable correspondiente principalmente a las hojas de la hélice y algunas hélices que componen el dominio catalítico. Los colores cercanos al verde representan regiones intermedias en relación con la capacidad y el rango de movimiento. Sobre la base de esta información, es posible percibir esta tinción presente en bucles en los extremos de los modelos, así como en algunas hélices del dominio catalítico. Por último, están las regiones que presentaron una coloración naranja/roja, en la que se representa un enorme potencial de movimiento. Por lo tanto, se puede ver que hay pequeñas regiones de bucles en los extremos de los dominios catalíticos con este potencial de movimiento, así como una hélice situada en la porción del dominio catalítico, más específicamente delante de la tríada catalítica de los OPB, en la que obtuvo el movimiento más expresivo del estudio.
Todos los modelos obtuvieron la misma configuración de movimiento, donde una hélice (en naranja) demostró ser la región con la mayor capacidad de movimiento. Así, los OPB de L. brasiliensis, L. donovani, L. infantum, L. mexicana y L. panamensis en su mayor rango de movimiento obtenidos, respectivamente, 9,8o, 11,3o, 11,8o, 11,9o y 11,6o (figuras 12 y 13).
Figura 12: Resultado del análisis por modos normales en representación por el programa pymol, en la imagen se evidencian los movimientos de los modelos, en sus formas relajadas (derecha) en comparación con el movimiento de mayor amplitud (izquierda). También se representan las regiones más rígidas (azul oscuro), regiones con poco movimiento (azul claro), regiones intermedias (verde), regiones con buen potencial de movimiento (naranja) y regiones extremadamente maleables (rojo) (Parte1/2).
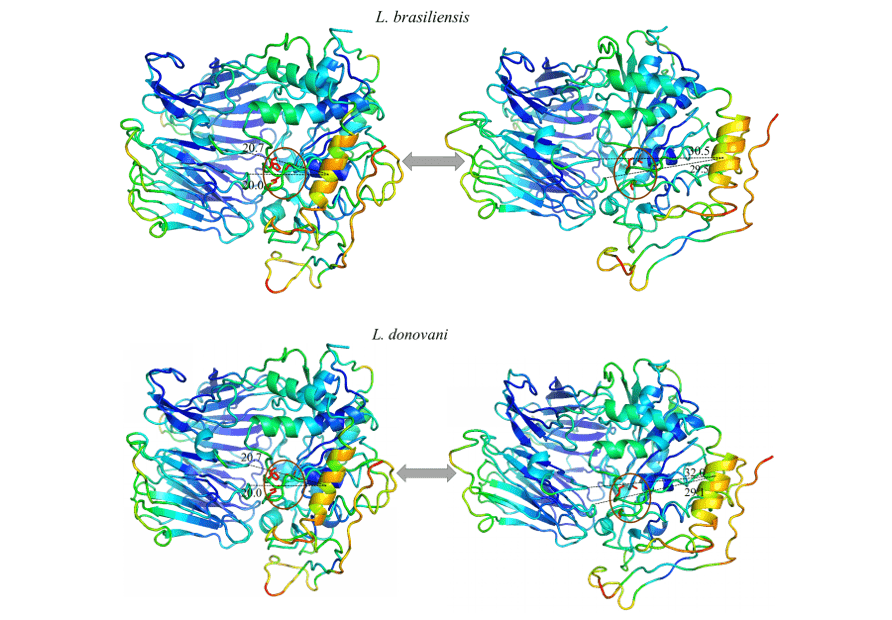
Figura 13: Resultado del análisis por modos normales en representación por el programa pymol, en la imagen se evidencian los movimientos de los modelos, en sus formas relajadas (derecha) en comparación con el movimiento de mayor amplitud (izquierda). También se representan las regiones más rígidas (azul oscuro), regiones con poco movimiento (azul claro), regiones intermedias (verde), regiones con buen potencial de movimiento (naranja) y regiones extremadamente maleables (rojo). (Parte 2/2)
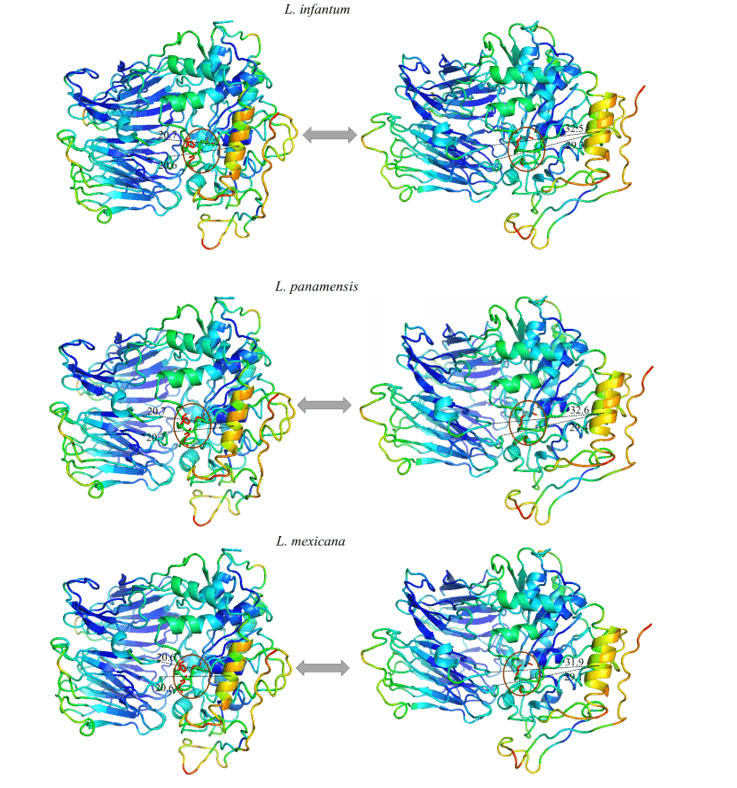
En vista de esto, los estudios normales respaldan un posible mecanismo de OPBs aún no descrito para las especies de Leishmania spp. hasta el presente estudio.
CONCLUSIÓN
En este estudio, se obtuvieron los modelos tridimensionales de la enzima opb de L.brasiliensis, L. donovani, L. infantum, L. mexicana y L. panamensis. La validación de los modelos presentó resultados fiables para todos los modelos tridimensionales obtenidos. En la caracterización de la enzima, el mapa de potencial electrostático de la superficie mostró que la mayoría de los residuos presentaban carga negativa. En la caracterización de la región alrededor de la tríada catalítica, demostró similitud entre el volumen, el área y la correspondencia entre los residuos positivos y negativos. Por lo tanto, fue posible verificar que los resultados del análisis por modos normales sugestivan un movimiento expresivo en una de hélice específica, que se produjo una distancia lineal de esta, desde el centro hacia la periferia, exponiendo así la tríada catalítica. La descripción de estos movimientos realizados por esta enzima es de gran importancia para ayudar a la comprensión de su funcionamiento.
Por último, los resultados del presente estudio pueden añadir conocimiento a la comunidad científica, aportando aclaraciones y nuevas preguntas relacionadas con el tema, sirviendo de base para estudios eventuales en el área de la salud.
REFERENCIAS
A. Benner S, Cannarozzi G, Gerloff D, Turcotte M, Chelvanayagam G. Bona Fide Predictions of Protein Secondary Structure Using Transparent Analyses of Multiple Sequence Alignments. Chem Rev. 1997;97(8):2725-2844. doi:10.1021/cr940469a.
Alva, V., Nam, S. Z., Söding, J., & Lupas, A. N. (2016). The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Research, 44(W1), W410–W415. doi.org/10.1093/nar/gkw348.
Alvarenga DG, Escalda PMF, da Costa ASV, Monreal MTFD. Leishmaniose visceral: Estudo retrospectivo de fatores associados à letalidade. Rev Soc Bras Med Trop. 2010;43(2):194-197.
Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389‐3402. doi:10.1093/nar/25.17.3389.
Andersson CD, Chen BY, Linusson A. Mapping of ligand-binding cavities in proteins [published correction appears in Proteins. 2011 Apr;79(4):1363]. Proteins. 2010;78(6):1408–1422. doi:10.1002/prot.22655.
Bailey F, Mondragon-Shem K, Hotez P, et al. A new perspective on cutaneous leishmaniasis-Implications for global prevalence and burden of disease estimates. PLoS Negl Trop Dis. 2017;11(8):e0005739. Published 2017 Aug 10. doi:10.1371/journal.pntd.0005739.
Carmo RF, Luz ZMP da, Bevilacqua PD. Percepções da população e de profissionais de saúde sobre a leishmaniose visceral. Cien Saude Colet. 2016;21(2):621-628. doi:10.1590/1413-81232015212.10422015.
Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5(4):823‐826.
Derewenda ZS, Derewenda U, Kobos PM. (His)Cε-H···O=C< Hydrogen Bond in the Active Sites of Serine Hydrolases. J Mol Biol. 1994;241(1):83-93. doi:10.1006/JMBI.1994.1475.
Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32(Web Server issue):W665–W667. doi:10.1093/nar/gkh381.
Eisenberg, D., Lüthy, R., & Bowie, J. U. (1997). [20]VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods in Enzymology, 277, 396–404. https://doi.org/10.1016/S0076-6879(97)77022-8.
Eyal E, Lum G, Bahar I. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics. 2015;31(9):1487–1489. doi:10.1093/bioinformatics/btu847.
Ghorbani Masoud, Farhoudi Ramin. Leishmaniasis in humans: drug or vaccine therapy? Drug Des Devel Ther. 2018;12:25-40. doi:10.2147/DDDT.S146521.
Hedstrom L. Serine Protease Mechanism and Specificity. Chem Rev. 2002;102(12):4501-4524. doi:10.1021/cr000033x.
Katsila T, Spyroulias GA, Patrinos GP, Matsoukas MT. Computational approaches in target identification and drug discovery. Comput Struct Biotechnol J. 2016;14:177–184. Published 2016 May 7. doi:10.1016/j.csbj.2016.04.004.
Kumar S, Tamura K, Nei M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5(2):150-163.
Macedo-Silva RM, dos Santos C de LP, Diniz VA, De Carvalho JJ, Guerra C, Côrte-Real S. Peripheral blood fibrocytes: New information to explain the dynamics of Leishmania infection. Mem Inst Oswaldo Cruz. 2014;109(1):61-69. doi:10.1590/0074-0276130247
Machado P de A, Carneiro MPD, Sousa-Batista A de J, et al. Leishmanicidal therapy targeted to parasite proteases. Life Sci. 2019;219:163-181. doi:10.1016/J.LFS.2019.01.015.
Morris, A. L., MacArthur, M. W., Hutchinson, E. G., & Thornton, J. M. (1992). Stereochemical quality of protein structure coordinates. Proteins: Structure, Function, and Bioinformatics, 12(4), 345–364. https://doi.org/10.1002/prot.340120407.
Ovchinnikova M V., Mikhailova AG, Karlinsky DM, Gorlenko VA, Rumsh LD. Reversible cyclic thermal inactivation of oligopeptidase B from Serratia proteamaculans. Acta Naturae. 2018;10(2):65-70.
Ramachandran, G. N., Ramakrishnan, C., & Sasisekharan, V. (1963). Stereochemistry of polypeptide chain configurations. Journal of Molecular Biology, 7(1), 95–99. https://doi.org/10.1016/S0022-2836(63)80023-6.
Santos Filho, O. A., & Alencastro, R. B. de. (2003). Modelagem de proteínas por homologia. Química Nova, 26(2), 253–259. https://doi.org/10.1590/S0100-40422003000200019
Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. Published 2011 Oct 11. doi:10.1038/msb.2011.75.
SODERO ACR, DOS SANTOS ACGO, MELLO JFRE, et al. Oligopeptidase B and B2: comparative modelling and virtual screening as searching tools for new antileishmanial compounds. Parasitology. 2017;144(4):536-545. doi:10.1017/s0031182016002237.
Swenerton RK, Zhang S, Sajid M, et al. The oligopeptidase B of Leishmania regulates parasite enolase and immune evasion. J Biol Chem. 2011;286(1):429-440. doi:10.1074/jbc.M110.138313.
Volkamer, A., Kuhn, D., Rippmann, F., & Rarey, M. (2012). Dogsitescorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics, 28(15), 2074–2075. https://doi.org/10.1093/bioinformatics/bts310.
Wang Q, Arighi CN, King BL, et al. Community annotation and bioinformatics workforce development in concert–Little Skate Genome Annotation Workshops and Jamborees. Database (Oxford). 2012;2012:bar064. Published 2012 Mar 20. doi:10.1093/database/bar064
Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics. 2016;54:5.6.1–5.6.37. Published 2016 Jun 20. doi:10.1002/cpbi.3.
Wiederstein, M., & Sippl, M. J. (2007). ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research, 35(SUPPL.2), 407–410. https://doi.org/10.1093/nar/gkm290.
WHO. Integrating Neglected Tropical Diseases into Global Health and Development: Fourth WHO Report on Neglected Tropical Diseases.; 2017. http://apps.who.int/iris/bitstream/10665/255011/1/9789241565448-eng.pdf?ua=1.
WHO. (2019). Leishmanioses – Informe Epidemiológico das Américas No 7 – Março, 2019. Retrieved from http://iris.paho.org/xmlui/bitstream/handle/123456789/50505/ 2019-cde-leish-informe-epi-das-americas.pdf?sequence=2&isAllowed=y.
ANEXO – FIGURAS Y TABLAS EN INGLÉS
Esquema 1: Esquema simplificado de pasos y métodos de material.
Tabla 1: Porcentaje de identidad entre los modelos de oligopeptity B de las especies de Leishmania y su respectivo moho.
| Mold protein (code PDB) | Models OPB
(código uniprot) |
Identity (%) | Gaps (%) |
| OPB
L. major (code PDB 2XE4) |
L.brasiliensis
(A4H5Q8) |
86 | 0 |
| L. donovani
(C9EF60) |
96 | 0 | |
| L. infantum
(A4HTZ8) |
96 | 0 | |
| L. Mexicana
(E9AMS8) |
90 | 0 | |
| L. panamensis
(A0A088RJA7) |
86 | 0 |
Fonte: Autoral.
Figura 6: Resultados de los gráficos ramachandran, obtenidos por el programa PROCHECK, las estructuras de los modelos DE OPB generados y el molde.
| Structures | % waste in the regions | ||||
| Favorable | Allowed | Unfavorable | |||
| OLIGOPEPTIDASE B | L. major (A)
(PDB 2XE4) |
90,2 | 9,5 | 0,3 | |
| L.brasilienses (B) | 92,2 | 7,7 | 0,2 | ||
| L. donovani (C) | 91,9 | 8,0 | 0,2 | ||
| L. infantum (D) | 92,3 | 7,3 | 0,3 | ||
| L. Mexicana (E) | 91,2 | 8,3 | 0,5 | ||
| L. panamensis (F) | 91,4 | 8,3 | 0,3 | ||
Fuente: Preparado por el autor en base a los resultados de procheck.
Figura 7: Resultados de la puntuación Z calculados en el servidor Web ProSA de las estructuras de molde (para la comparación). (A) L .major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. La región en azul oscuro indica la puntuación de las proteínas obtenidas por RMN y en azul claro de las proteínas obtenidas por difracción de rayos X
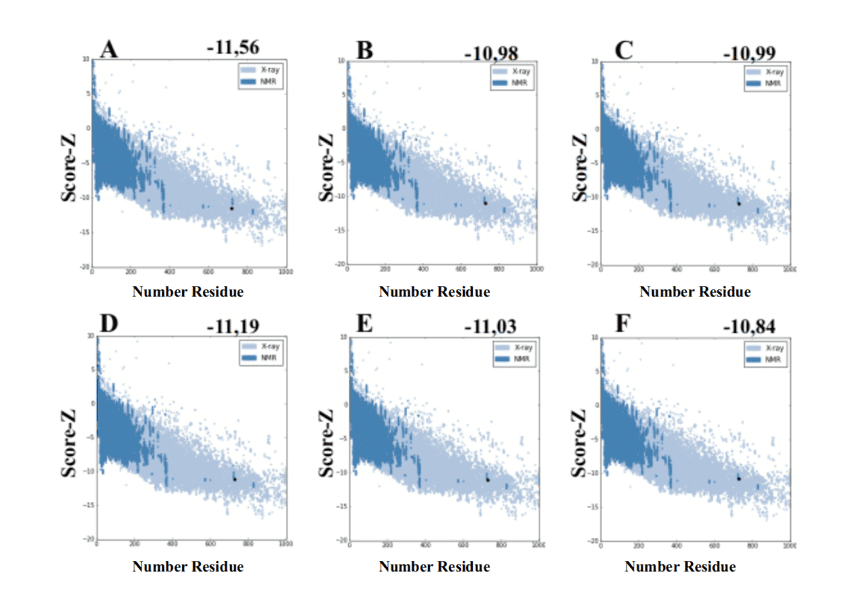
Tabla 2: Resultados de Verificar 3D, mostrando el porcentaje de residuos con puntuación > 0.2.
| Structures | % residue with score > 0,2 | |
| OLIGOPEPTIDASE B | L.major
(PDB 2XE4) |
93,20 |
| L.brasilienses | 95,62 | |
| L.donovani | 97,12 | |
| L.infantum | 94,93 | |
| L.mexicana | 95,62 | |
| L.panamensis | 94,93 |
Fuente: Autor.
Tabla 3: Comparación entre las estructuras secundarias predichas por psipred y las encontradas a través de Pymol.
| Models | α-Hélix | Sheet β | PSIPRED
α-Hélix |
PSIPRED
Sheet β |
| L.brasiliensis | 15 | 38 | 11 | 36 |
| L. donovani | 16 | 38 | 11 | 36 |
| L. infantum | 16 | 38 | 11 | 36 |
| L. mexicana | 15 | 38 | 10 | 36 |
| L. panamensis | 15 | 38 | 11 | 36 |
Fuente: Autor.
Tabla 4: RMSD de los OPB generados por el Modelador, teniendo con orientación los carbonos alfa del molde OPB de L. mayor.
| Mold | Models | RMSD (Å) |
| L. major
(2XE4) |
L.brasiliensis | 0,15 |
| L. donovani | 0,15 | |
| L. infantum | 0,16 | |
| L. mexicana | 0,19 | |
| L. panamensis | 0,14 |
Fuente: Autor.
Tabla 5: Valores que hacen referencia a las posibles regiones de conexión de los OPB (obtenidos por el servidor).
| Structures | DogSiteScoore | |||
| Volume | Area | Drug Score | ||
| OP OLIGOPEPTIDASE B | L. major
(PDB 2XE4) |
1690,62 | 1818,41 | 0,80 |
| L.brasiliensis | 1527,84 | 1766,63 | 0,80 | |
| L. donovani | 1074,57 | 1428,55 | 0,79 | |
| L. infantum | 1309,97 | 1572,19 | 0,80 | |
| L. mexicana | 800,92 | 799,38 | 0,85 | |
| L. panamensis | 971,96 | 1083,40 | 0,81 | |
Fuente: Autor.
[1] Biomédica y máster en Bioquímica Médica.
[2] Máster en Ciencias Farmacéuticas y Farmacéuticas por la UFRJ.
[3] Doctor en Química, Máster en Química Orgánica y Farmacéutica Industrial.
Enviado: Mayo, 2020.
Aprobado: Mayo, 2020.