ARTICOLO ORIGINALE
RIBEIRO, Fernando de Sá [1], JESUS, Jéssica Barbosa de [2], SOUZA, Alessandra Mendonça Teles de [3]
RIBEIRO, Fernando de Sá. JESUS, Jéssica Barbosa de. SOUZA, Alessandra Mendonça Teles de. Analisi e caratterizzazione di un promettente obiettivo terapeutico identificato nella Leishmania spp. Revista Científica Multidisciplinar Núcleo do Conhecimento. Ano 05, Ed. 05, Vol. 09, pp. 99-132. Maio de 2020. ISSN: 2448-0959, Collegamento di accesso: https://www.nucleodoconhecimento.com.br/salute/bersaglio-terapeutico
RIEPILOGO
La leishmaniosi è una malattia trascurata causata dai protozoi del genere Leishmania spp., che colpisce circa 1,6 milioni di individui ogni anno e 500.000 si presentano nella forma viscerale. In Brasile ci sono circa 30.000 nuovi casi ogni anno. Inoltre, il paese è responsabile del 90% dei casi segnalati di leishmaniosi viscerale, e questa è la forma più grave della malattia. Alleato a questi fatti, il trattamento attuale è inefficace, contribuendo alla creazione di ceppi resistenti. Attualmente, il trattamento ha diversi effetti collaterali e danni permanenti alla salute dei pazienti, questo fatto ha contribuito alla ricerca di nuovi farmaci contro la leishmaniosi. L’enzima oligopeptidase B (OPB) è stato studiato come possibile bersaglio terapeutico nello sviluppo di agenti antiparassiti. Così, l’obiettivo di questo lavoro è quello di costruire il modello tridimensionale dell’enzima Oligopeptidase B di diverse specie di Leishmania spp. e confrontarli tra loro. A tal fine, è stato utilizzato il metodo di modellazione comparativa. In questo metodo, i modelli della specie L. brasiliensis, L. donovani, L. infantum, L. mexicana e L. panamensis sono stati costruiti utilizzando il programma MODELLER. Una volta che i modelli erano pronti, il processo di convalida è stato effettuato e successivamente caratterizzato, che è stato possibile verificare un promettente grado di somiglianza tra i modelli. Infine, questi modelli sono stati sottoposti al metodo di analisi da modalità normali, che ha ottenuto un modello di movimento simile, quindi è stato possibile verificare un movimento in una regione specifica di un alfa-elica, portando di conseguenza alla triade dell’enzima esposto, che può essere indicativo di un meccanismo di azione. Infine, si prevede di utilizzare i modelli costruiti per assistere nello sviluppo di una nuova terapia promettente per il trattamento della leishmaniosi.
Parole chiave: Leishmaniosi, oligopeptidase B, modellazione molecolare, modalità normali.
INTRODUZIONE
La leishmaniosi, causata dalla leishmania spp., è una malattia caratterizzata da diversi tipi di manifestazioni, da lievi, in cui ci sono segnalazioni di piccole lesioni che anche senza il dovuto trattamento regrediscono a quelle più gravi come la Leishmaniosi Viscerale (VL). In Brasile, la forma più grave di questa malattia presenta dati allarmanti rispetto ad altri paesi, rendendo il paese il più grande detentore di casi di VL in tutta l’America (ALVARENGA, 2010; CHI, 2019).
Questa malattia appartiene al gruppo di malattie trascurate, che fanno parte di tutte quelle malattie che colpiscono principalmente i paesi sottosviluppati e le regioni più povere, in modo che non suscitano interesse nello sviluppo di farmaci. Pertanto, è necessario che tecniche efficienti e a basso costo sopravvivano per superare questa mancanza di incentivi finanziari per lo studio di questa malattia. Così, metodi computazionali possono essere utilizzati al fine di ridurre il tempo nello sviluppo di una nuova terapia promettente e di conseguenza il costo rispetto ai metodi più tradizionali per lo sviluppo di farmaci (BAILEY et al., 2017; CHI, 2017).
Nonostante il poco investimento in questo settore, c’è un trattamento per la malattia come le pente valenteanti antimoniali (farmaco di prima scelta) o l’amphotericina B (farmaco di seconda scelta). Tuttavia, tali trattamenti presentano diversi svantaggi, come l’alto tasso di resistenza e l’ampia varietà di effetti collaterali, che vanno dal mal di mare ai possibili problemi causati alla terza (terza) coppia del nervo cranico, che porta a difficoltà motorie (MACEDO-SILVA et al., 2014).
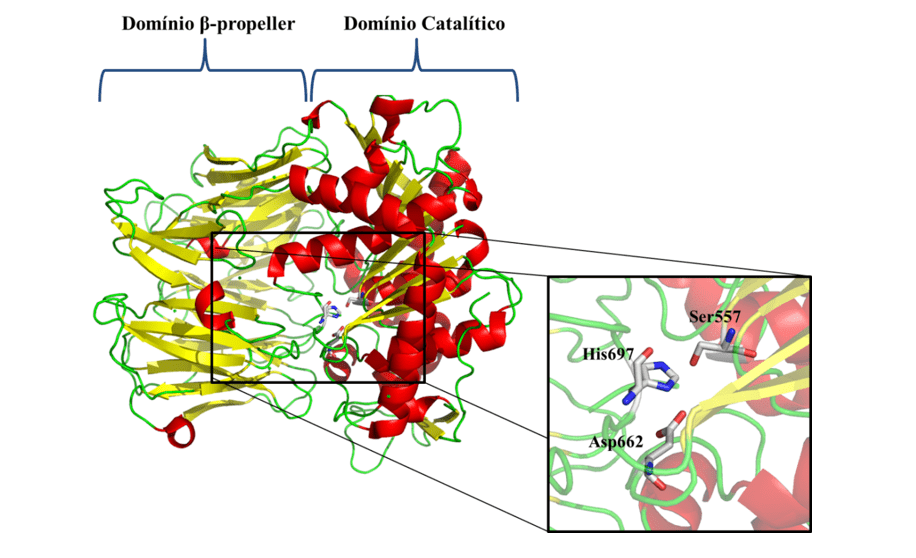
Alla luce dei problemi presentati e dell’importanza epidemiologica della leishmaniosi, è nota la necessità di sforare nuovi obiettivi terapeutici specifici contro la malattia. Come uno dei nuovi obiettivi terapeutici, abbiamo Oligopeptidase B (OPB) (Figura 1), che è una proteasi serina, appartenente alla sottofamiglia S9A, avendo come caratteristica una triade catalitica composta dai residui di aminoacidi serine (Ser), acido asaptico (Asp) e istidina (Sua) che si trovano tra i due domini. Questo enzima ha come caratteristica la capacità di fendere i residui delle strutture proteiche. Inoltre, è descritto nella letteratura come un componente chiave per il meccanismo di fuga immunitaria del parassita, fendendo enolasi, una proteina che opsonisar il protozoo, in modo che quando riconosciuto dal macrofago viene distrutto. Tuttavia, mentre il parassita è all’interno del macrofago, l’OPB è super espresso, causando il parassita non essere riconosciuto all’interno della cellula, quindi si moltiplicherà fino a quando non viene levigato (SODERO et al., 2016; SWENERTON et al., 2011; OVCHINNIKOVA et al. 2018).
Figura 1: struttura tridimensionale dell’OPB principale l. (codice PDB 2XE4) (MCLUSKEY et al., 2010) che mostra i domini catalitici e di eliche. Le strutture secondarie, come l’elica a z, le foglie e le maniglie, sono mostrate rispettivamente in rosso, giallo e verde. In particolare, vengono mostrati i residui (Ser557, Asp662 e His697) della triade catalitica.
Pertanto, lo studio in questione mirava a indagare e caratterizzare gli OBB e i loro siti attivi, della specie L. brasiliensis, L, donovani, L. infantum, L. mexicana e L. panamensis. Infine, ci si aspettava di identificare possibili somiglianze tra le proteine, in modo da consentire il futuro sviluppo di una nuova terapia promettente con un ampio spettro di azione sugli enzimi di tutte le specie nello studio.
OBIETTIVO GENERALE
In considerazione della necessità di sviluppare nuove entità chimiche per il trattamento contro la leishmaniosi, questo lavoro aveva come obiettivo principale quello di costruire e caratterizzare l’enzima oligopeptidase B di Leishmania spp. utilizzando tecniche di studio computazionale.
OBIETTIVI SPECIFICI
- Costruire e convalidare i modelli di oligopeptidasi B enzimi delle specie Leishmania;
- Eseguire la caratterizzazione degli enzimi;
- Eseguire studi di simulazione in base alle modalità normali.
MATERIALE E METODI
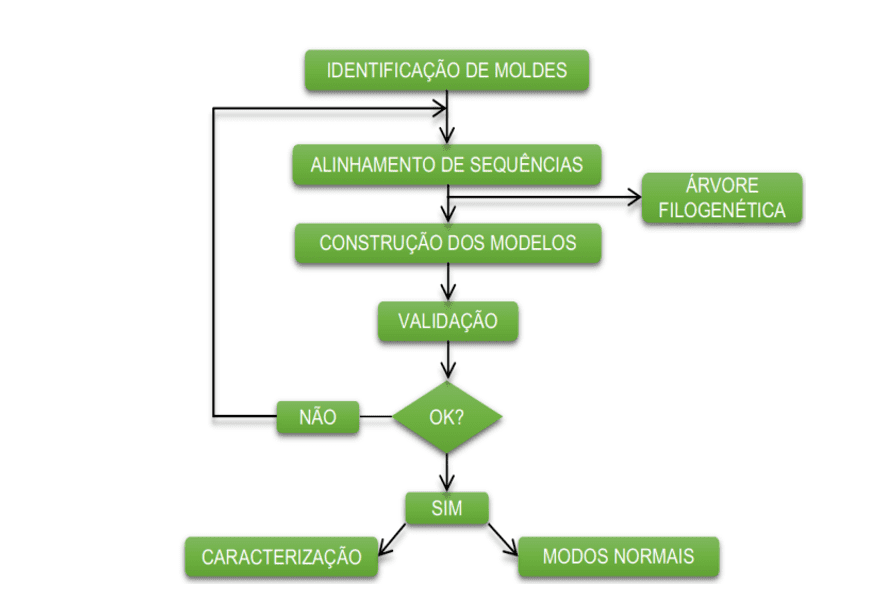
Il diagramma seguente (diagramma 1) mostra in modo semplificato i passaggi utilizzati durante lo sviluppo di questo lavoro.
Schema 1: schema semplificato di passaggi e metodi del materiale.
OTTENERE LA STRUTTURA PRIMARIA
Le strutture primarie degli OBB leishmanias sono state ottenute dal database Uniprot (The Universal Protein Resource) (WANG et al., 2012).
ID STAMPO
Quindi, la ricerca delle proteine dello stampo è stata eseguita dall’allineamento tra la sequenza di amminoacidi delle proteine bersaglio e le sequenze di proteine amminoacidi depositate nella Protein Data Bank (PDB) dal server BLAST (Basic Local Alignment Search Tool) (ATLSCHUL et al., 1997) in base all’identità tra sequenze di amminoacidi.
ALIGNMENT DI SEQUENZI
Una volta definito lo stampo da utilizzare, la sequenza di stampi è stata allineata con i rispettivi OBB, nel programma ClustalOmega (ClustaO) (SIEVERS et al., 2011).
COSTRUZIONE DEL MODELLO
Dopo aver ottenuto l’allineamento, queste informazioni sono state utilizzate nel programma MODELLER v9.20 per costruire le strutture 3D dei modelli (WEBB e SALI, 2016).
CONVALIDA DEI MODELLI COSTRUITI
Per la convalida dei modelli, è stato utilizzato il grafico Ramachandran generato dal server PDBsum, Verify 3D e ProSA-web, tutti ottenuti dal server Saves.
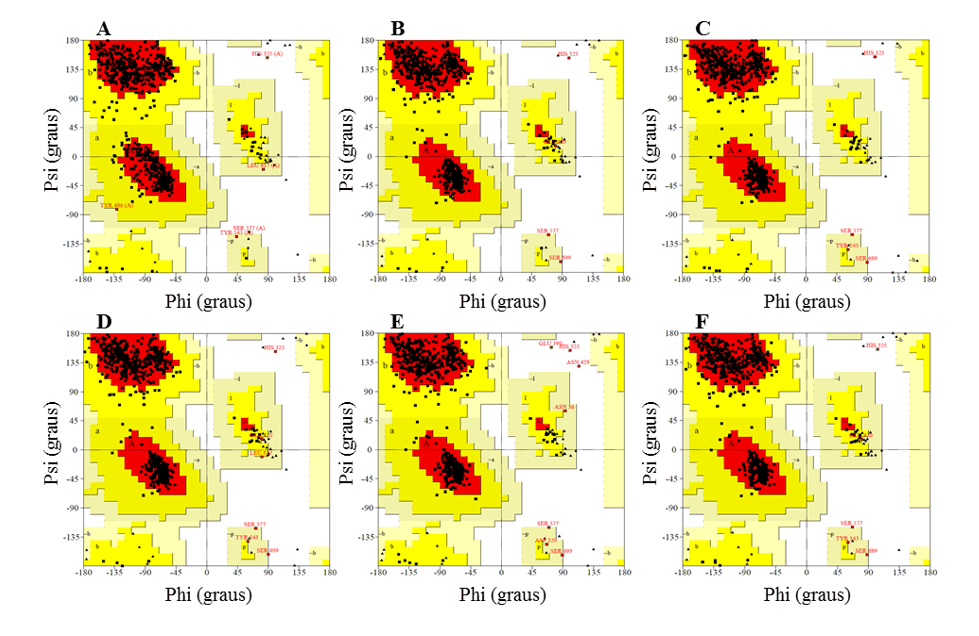
Nell’analisi del grafico di Ramachandran, è consentito visualizzare gli angoli diedrici (phi ( ) e psi (z)) dei residui di amminoacidi nella struttura delle proteine. L’angolo phi è il risultato del legame tra il gruppo NH e il carbonio alfa, mentre l’angolo psi (z) deriva dal legame tra il carbonio alfa e il gruppo carbonilico (RAMACHANDRAN et al., 1963).
Il grafico fornisce un modo semplice per visualizzare la distribuzione degli angoli torm di una struttura proteica. Inoltre, fornisce una panoramica delle regioni consentite e non consentite dei valori dell’angolo torm, che fungono da fattore importante nella valutazione della qualità delle strutture proteiche 3D. Con questo, definisce i residui che si trovano nelle regioni che sono energeticamente più favorevoli e sfavorevoli e guida la valutazione della qualità dei modelli teorici o sperimentali delle proteine. Questo grafico è diviso in regioni, in modo che le regioni più favorevoli siano in rosso, regioni aggiuntive più permissive in regioni marroni, permissive in bianco e non permissive. Secondo questa convalida per un modello previsto da considerare di ottima qualità, deve avere più del 90% dei residui di amminoacidi nella regione più favorevole (SANTOS-FILHO e ALENCASTRO, 2003).
Inoltre, deve avere la maggior parte dei suoi residui nelle regioni più favorevoli, oltre a non avere residui nelle regioni non permissive, ad eccezione degli amminoacidi glicina (Gly) e proline (Pro) che sono eccezioni per questa zona. Questi due residui presentano variazioni nella catena laterale che conferiscono una maggiore rigidità, nel caso di Pro, e una maggiore flessibilità, nel caso di Gly, così può assumere angolazioni inaspettate. Per questo motivo sono accettati nelle regioni non permissive del grafico. L’esistenza delle regioni non ammesse è dovuta al fatto che ci sono effetti sterici tra i residui (catene laterali) di aminoacidi. (MORRIS et al., 1992).
Verifica 3D analizza la compatibilità di un modello atomico (3D) con la propria sequenza di aminoacidi (1D). Ogni residuo riceve una classe strutturale in base alla sua posizione e ambiente (alfa, beta, loop, polare, apolar, ecc.). Poco dopo, un database generato da buone strutture viene utilizzato per ottenere un punteggio per ciascuno dei 20 aminoacidi in questa classe strutturale. L’asse verticale sul grafico rappresenta il punteggio medio del profilo 3D-1D per ogni residuo in una finestra scorrevole di 21 residui e i risultati sotto forma di punteggi vanno da -1 (punteggio scarso) a 1 (buon punteggio) (EISENBERG et al., 1997).
ProSa-web calcola un punteggio di qualità complessivo per una struttura di input specifica. Se il punteggio di questo è al di fuori di una caratteristica di gamma per le proteine native, la struttura probabilmente contiene errori. Molti punteggi di qualità locali indicano parti problematiche del modello che sono evidenziate anche in un visualizzatore di molecole 3D per una facile rilevazione (WIEDERSTEIN e SIPPL, 2007).
ANALISI DELLA STRUTTURA SECONDARIA
Dopo la fase di convalida, le strutture terziarie dei modelli sono state confrontate con la distribuzione della struttura secondaria prevista da Quick2D, disponibile sul server Bioinformatics Toolkit (https://toolkit.tuebingen.mpg.de/), scegliendo così i modelli (ALVA et al., 2016).
CHARACTERIE DI OLIGOPEPTIDASES (OPBS) DI LEISHMANIAS
MAPPA POTENZIALE ELETTROSTATICA (MEP)
Per ottenere gli eurodeputati delle superfici e dei siti di collegamento degli OBB, è stata utilizzata un’estensione del programma PyMOL, strumenti apb (BAKER et al., 2001). Prima di essere analizzati in PyMOL, gli enzimi sono stati preparati nel Server PDB2PQR (http://nbcr-222.ucsd.edu/pdb2pqr_2.0.0/) utilizzando il parametro standard del server (DOLINSKY et al., 2004).
CARATTERIZZAZIONE DI SITI OPBS E SITI SECONDARI
I modelli e lo stampo sono stati presentati nella piattaforma proteina plus utilizzando l‘opzione Dogsitescorer (VOLKAMER et al., 2012), in cui le cavità sono state previste nelle strutture 3D dei modelli. Ciò ha generato risultati relativi a possibili siti di legame e siti secondari enzimatici per la previsione di queste cavità. Il programma si avvale di una mesh tridimensionale il cui bordo può essere regolato tra 0,2 e 1,0 , insieme a un filtro gaussiano che viene utilizzato per identificare le cavità sulla superficie della proteina che sono adatte ad ospitare gli atomi di ligando. Inoltre, DoGSiteScorer prevede anche la drogabilità per ogni cavità prevista. Così, per ogni interazione tra la proteina e il possibile farmaco, viene assegnato un punteggio che si riferisce alla droga della cavità, chiamata drugscore. Per stimare il valore di drugscore, il programma utilizza una macchina vettoriale di supporto (SVM), in cui vengono utilizzati i seguenti descrittori: volume, proporzione di residui non polari e profondità (VOLKAMER et al., 2012).
ALBERO FILOGENETICO
Infine, il grado di parentela evolutiva tra le specie di Leishmanie è stato analizzato utilizzando il programma MEGA (Molecular evulutionary genetics analysis) program, utilizzando i metodi di giunzione del vicino, che consente la costruzione dell’albero filogenetico al fine di definire le prossimità evolutive tra popolazioni di sequenze precedentemente definite dall’utente (KUMAR et al., 2004).
MODALITÀ NORMALE
Per eseguire le modalità normali, sono stati utilizzati i file generati nelle fasi di energia minimizaes realizzati da GROMACs versione 5.1.2. Sono stati eseguiti i primi 4 passi relativi al processo di dinamica molecolare. Il primo è stata la generazione di file di topologia, con l’aggiunta di idrogeno alla proteina. Nel secondo, è stata creata la scatola dell’acqua, e questo è un passo molto importante per il calcolo dell’interazione tra la proteina e il solvente. Nel terzo, gli ioni sono stati aggiunti per stabilire un sistema neutrale. Infine, nella quarta fase, sono state eseguite miniminizze energetiche, in cui è stato inserito il campo di forza AMBER99SB. Da questo punto in poi, le modalità normali dei modelli sono state eseguite utilizzando il server ANM (Anisotropic Network Model) (http://anm.csb.pitt.edu/) al fine di analizzare il movimento degli enzimi delle specie Leishmania e anche per osservare alcune caratteristiche importanti per gli enzimi, come i possibili movimenti legati al meccanismo d’azione (EYAL et al., 2015).
RISULTATI E DISCUSSIONI
SELECTION DI PROTEINE MOLD E ALIGNMENT BETWEEN SEQUENZI
Abbiamo ottenuto 100 strutture primarie degli OBB di leishmania spp. utilizzando il server UniProt, con 5 specie di Leishmanias selezionate. Le sequenze di amminoacidi riviste selezionate dall’enzima OPB sono state L.brasiliensis, L. donovani, L. infantum, L. mexicana e L. panamensis sotto i codici A4H5Q8, C9EF60, A4HT-8, E9AMS8 e A0A088RJA7, rispettivamente Queste specie sono state selezionate, a causa della loro elevata incidenza in Sud America e della loro resistenza contro l’attuale trattamento per la leishmaniasis (GHORBanI, 201).
Per la costruzione di modelli 3D, il programma BLAST è stato utilizzato per confrontare le sequenze di amminoacidi delle sequenze bersaglio con sequenze proteiche di strutture tridimensionali chiarite sperimentalmente. Sulla base dell’identità tra le sequenze e il numero di lacune, l’OPB ENZYME di L. major (codice PDB 2XE4) è stato selezionato come proteina di stampo (MCLUSKEY et al., 2010). L’identità tra gli enzimi bersaglio e il rispettivo stampo presentava valori compresi tra l’86% e il 96% (tabella 1). La percentuale di identità tra due sequenze si riferisce alla presenza dello stesso aminoacido nella stessa posizione tra le sequenze allineate. Per la costruzione di un modello di una proteina con più di 80 residui di aminoacidi, la percentuale di identità tra le strutture primarie dello stampo e il modello dovrebbe essere superiore al 25%. Inoltre, la percentuale di divari deve essere bassa del 20% per essere considerata un buon allineamento (SANTOS-FILHO e ALENCASTRO, 2003). Pertanto, la probabilità di somiglianza delle strutture tridimensionali delle proteine è alta.
Tabella 1: Percentuale di identità tra i modelli di oligopeptity B delle specie di Leishmania e la loro rispettiva muffa.
| Proteína molde (código PDB) | Modelos de OPB
(código uniprot) |
Identidade (%) | Gaps (%) |
| OPB
L. major (código PDB 2XE4) |
L.brasiliensis
(A4H5Q8) |
86 | 0 |
| L. donovani
(C9EF60) |
96 | 0 | |
| L. infantum
(A4HTZ8) |
96 | 0 | |
| L. Mexicana
(E9AMS8) |
90 | 0 | |
| L. panamensis
(A0A088RJA7) |
86 | 0 |
Fonte: Autore.
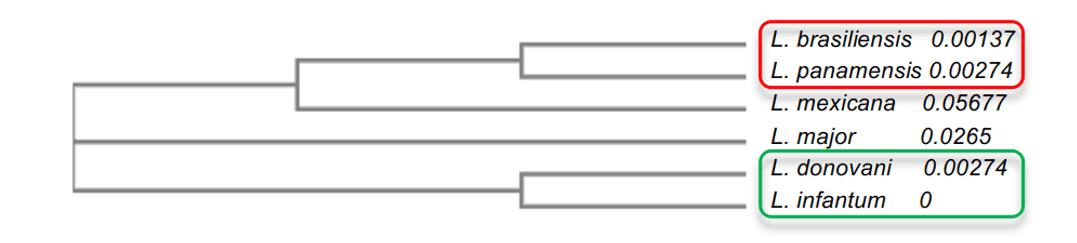
Nell’analisi filogenetica degli alberi, è stato possibile spiegare il grado di identità tra le specie di leishmania. È stato osservato che le specie con un più alto grado di identità, come L. infantum e L. donovani, entrambe con il 96%, presentavano una vicinanza l’una all’altra, oltre ad essere più vicine a L.major. Le altre specie presentavano valori più bassi come L.mexicana (90%), che era in mediana rispetto agli altri modelli. Infine, i modelli con la più bassa percentuale di identità, L. brasiliensis e L. panamensis, entrambi con l’86%, erano evolutivamente più distanti dal loro stampo, ma erano molto vicini l’uno all’altro (Figura 2). Questa analisi ha permesso di comprendere la differenza tra il grado di identità tra le specie e la comprensione di alcune importanti caratteristiche dell’enzima tra le specie.
Figura 2: Schema di alberi filogenetici della Leishmania spp. enzimi OBB. In questa rappresentazione, sono evidenziati i gruppi con la più alta somiglianza tra loro, dove il gruppo 1 è evidenziato in rosso e il gruppo 2 in verde.
MODELLI TRIDIMENSIONALI DELL’ENZIMA DI LEISHMANIA SPECIES
I modelli 3D della specie sono stati costruiti utilizzando il programma Modeller (WEBB e SALI, 2016) dall’allineamento delle strutture primarie (figura 3 e 4).
Figura 3: Risultato dell’allineamento tra le sequenze primarie degli OBB selezionati in UNIPROT (Parte 1/2).

Figura 4: Risultato dell’allineamento tra le sequenze primarie degli OBB selezionati in UNIPROT (Parte 2/2).

Osservando il risultato dell’allineamento delle strutture (Figura 3 e 4), è stato osservato un più alto grado di somiglianza tra L. donovani e L. infantum (gruppo 2) e una maggiore corrispondenza tra L. brasiliensis e L. panamensis (gruppo 1). Questi risultati rafforzano ciò che è stato presentato dall’albero filogenetico nella Figura 2, dove si suggerisce una vicinanza tra la specie, L. donovani e L. infantum e un’altra vicinanza tra la specie di L. brasiliensis e L. panamensis. D’altra parte, i due gruppi menzionati presentavano una maggiore distanza evolutiva rispetto agli altri. Questo fatto è noto quando viene effettuato un confronto nell’analisi di allineamento, dove le strutture primarie hanno mostrato una maggiore differenza tra i residui, rispetto ai due gruppi. Inoltre, il risultato dell’allineamento di L. mexicana si è rivelato molto promettente, il che si riferisce al sostegno di questa discussione proposta, poiché le differenze osservate rispetto alle altre specie erano equivalenti, a volte al primo gruppo citato, a volte al secondo. In alcune parti della sequenza analizzata, sono state osservate anche mutazioni specifiche che sono caratteristiche solo di L. mexicana in alcune parti della sequenza analizzata. Così, questo fatto può essere attestato a causa della sua posizione nella mediana nell’albero filogenetico, in relazione alle altre specie.
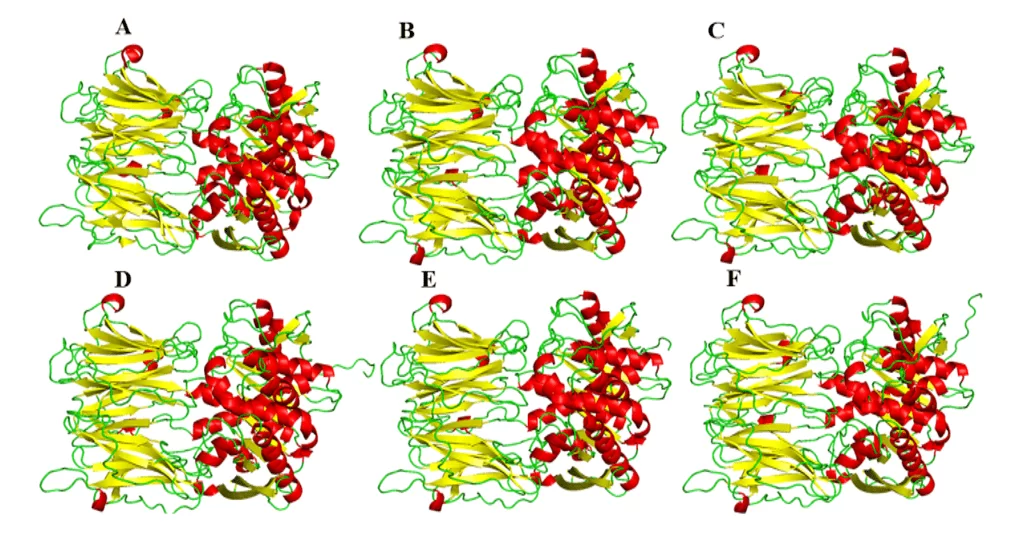
Come risultato della modellazione comparativa, sono stati ottenuti i modelli del presente studio, che ha suggerito una grande somiglianza visiva nella sua forma tridimensionale in relazione allo stampo (Figura 5). Per quanto riguarda il confronto tra i modelli, essi presentavano tra loro diverse quantità nelle strutture di z-helix e foglia-z. Questa differenza può essere spiegata dalle differenze specifiche nella composizione dei residui proteici, ma tali differenze non si presentavano in importanti regioni di questi enzimi, come il sito di legame. Pertanto, queste differenze non sono così importanti da modificare le strutture o il profilo di interazione con un possibile farmaco nel sito di legame.
Figura 5: Modelli e stampi degli OBB di leishmania spp., essendo evidenziato in rosso le z-elica, verde le maniglie e giallo le foglie- Para OPB, (A) L.major (molde) e modelos: (B) L. brasiliensis, (C) L. donovani, (D) L. infantum, (E) L. mexicana e (F) L. panamensis.
Nella fase di convalida, sono stati analizzati sulla base dell’analisi del grafico ramachandran (fornita dal server PDBsum), del punteggio 3D-1D (fornito dal programma verify-3D) e del punteggio z (fornito dal server ProSA-web). I valori ottenuti per i modelli sono stati confrontati con quelli ottenuti per lo stampo.
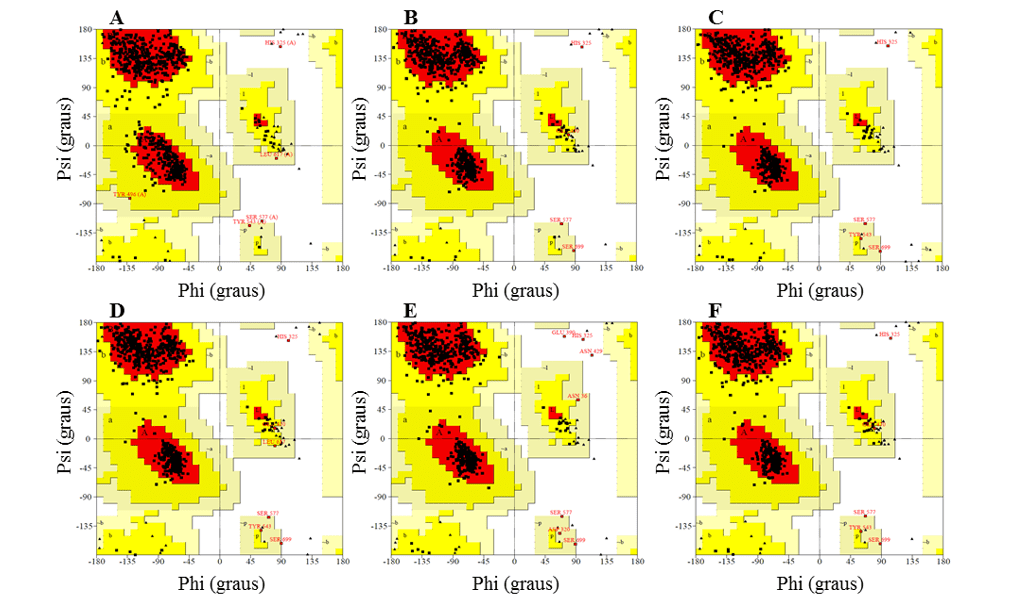
Nel grafico Ramachandran, i modelli presentavano la maggior parte dei residui nelle regioni favorevoli, che vanno tra il 91,2 e il 92,3%, mentre la percentuale di residui nelle regioni sfavorevoli era un massimo dello 0,5%, e i migliori modelli erano L.infantum e L.brasilienses. Questi modelli presentavano il maggior numero di residui nelle regioni favorevoli con il 92,3% e il 92,2% e la più bassa percentuale di residui nelle regioni sfavorevoli con rispettivamente lo 0,3% e lo 0,2% (Figura 6).
In tutti i modelli è stato possibile osservare che il residuo Ser, che fa parte della triade catalitica, si trovava nelle regioni sfavorevoli. Tuttavia, questo fatto non influisce sulla validità dei modelli, poiché quando si confronta lo stampo presentato lo stesso risultato. Pertanto, questo risultato non configura una bassa affidabilità dei modelli.
Figura 6: Risultati dei grafici ramachandran, ottenuti dal programma PROCHECK, dalle strutture dei modelli OBB generati e dallo stampo.
| Strutture | % di rifiuti nelle regioni | ||||
| Favorevole | Consentito | Sfavorevole | |||
| OLIGOPEPTIDASE B | L. major (A)
(PDB 2XE4) |
90,2 | 9,5 | 0,3 | |
| L.brasilienses (B) | 92,2 | 7,7 | 0,2 | ||
| L. donovani (C) | 91,9 | 8,0 | 0,2 | ||
| L. infantum (D) | 92,3 | 7,3 | 0,3 | ||
| L. Mexicana (E) | 91,2 | 8,3 | 0,5 | ||
| L. panamensis (F) | 91,4 | 8,3 | 0,3 | ||
Fonte: Preparato dall’autore in base ai risultati di procheck.
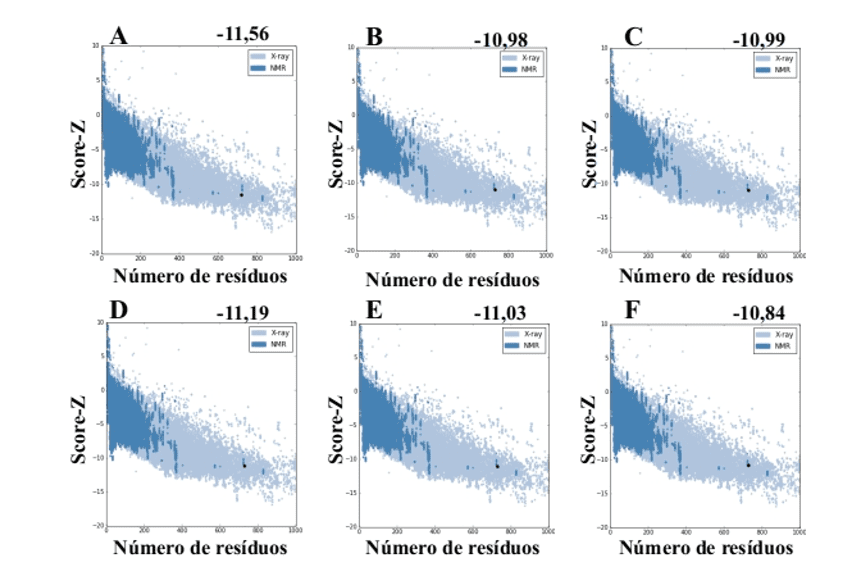
Quando si utilizza il server ProSA-web dai modelli generati da MODELLER, sono stati presentati i valori di punteggio da -10,84 a -11,19 e questi valori sono compatibili con le strutture PDB (Figura 7).
Figura 7: Risultati del punteggio z calcolati sul server ProSA-web delle strutture di stampo (per confronto). (A) L .major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. La regione in blu scuro indica il punteggio delle proteine ottenute dalla NMR e in azzurro delle proteine ottenute dalla diffrazione a raggi X.
Il programma Verify 3D è stato utilizzato per valutare la compatibilità tra le strutture 1D e 3D. I modelli ottenuti hanno presentato il 95,28% al 98,34% degli amminoacidi con compatibilità 3D-1D> 0.2, e i modelli L. donovani e L.brasilienses hanno ottenuto risultati migliori. Secondo i parametri ideali del programma, la maggior parte dei residui dovrebbe presentare valori superiori allo zero, poiché i valori al di sotto dello zero indicano regioni della molecola con problemi. La percentuale di amminoacidi con compatibilità 3D-1D deve essere superiore all’80% (EISENBERG et al., 1997). Pertanto, questi risultati indicano che i modelli presentavano compatibilità 1D-3D e i residui che presentavano l’incompatibilità non fanno parte del sito attivo degli enzimi (tabella 2).
Tabella 2: Risultati di Verifica 3D, che mostra la percentuale di residui con punteggio > 0.2.
| Estruturas | % de resíduos com score > 0,2 | |
| OLIGOPEPTIDASE B | L.major
(PDB 2XE4) |
93,20 |
| L.brasilienses | 95,62 | |
| L.donovani | 97,12 | |
| L.infantum | 94,93 | |
| L.mexicana | 95,62 | |
| L.panamensis | 94,93 |
Fonte: Autore.
VALIDATION DEL TRIAD CATALYTICO DELL’ENZIE
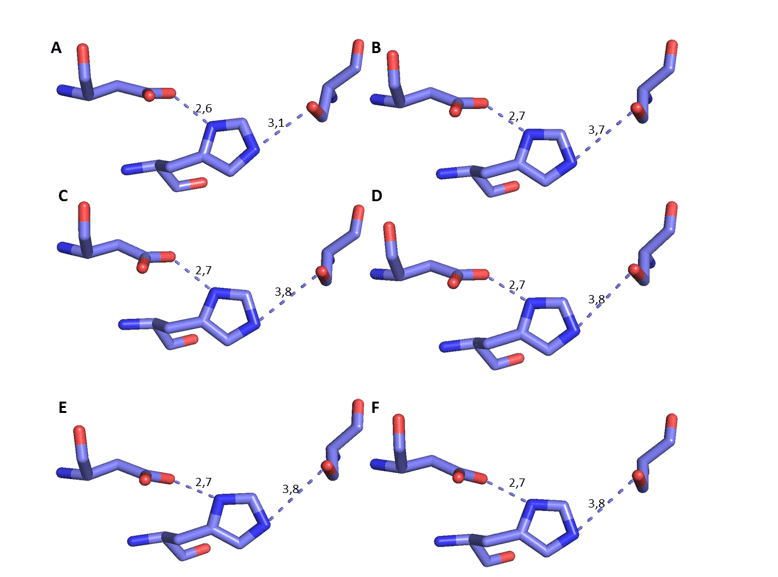
A causa del meccanismo di PB, è essenziale analizzare la distanza e l’orientamento tra i residui di aminoacidi e la triade catalitica (Ser, Asp e His) dei modelli generati al fine di aumentare l’affidabilità dei modelli.
È possibile osservare il confronto di questi residui specifici dei modelli OPB con lo stampo in questione nella figura 17. Inoltre, è stato possibile misurare la distanza tra i residui della triade catalitica, sulla base del meccanismo di interazione tra il sito e il substrato, con il Suo come riferimento (DEREWENDA et al. , 1994). Pertanto, ci deve essere una distanza specifica dal meccanismo di azione. Questa distanza dovrebbe essere di circa 3,5 % tra il suo ossigeno e l’azoto ser, oltre alla distanza di circa 2,6 s tra l’ossigeno aspartato e il suo altro azoto, come descritto nella letteratura (DEREWENDA et al. , 1994). In questa analisi, è stato possibile verificare che la distanza in questione abbia subito una piccola variazione ed è ancora conforme a quanto già descritto. Questa distanza è importante perché le proteasi serino richiedono la sua per scissione substrato (HEDSTROM, 2002) (Figura 8). Sulla base di questi risultati affidabili, è stata continuata un’ulteriore caratterizzazione dei modelli.
Figura 8: rappresentazione 3D della triade catalitica degli OBB, rispettivamente, Ser, His e Asp. Il colore blu scuro rappresenta l’atomo di azoto, l’ossigeno rosso e il carbonio lilla. Para OPB, (A) L.major (molde)e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis.
CHARACTERIE DI OLIGOPEPTIDASES DI LEISHMANIAS
STRUTTURA SECONDARIA DI STIMA
Dalla previsione della struttura secondaria degli enzimi oPP, sono stati rivelati il numero e la posizione delle strutture secondarie dei modelli. Il PSISPRED presentava tra 15 e 16 strutture z-helix e tutte presentavano 38 paia di foglie z (tabella 3). Dai risultati generati da PSIPRED, è stato eseguito un confronto visivo di questa struttura secondaria prevista con le strutture tridimensionali dei 5 modelli, utilizzando il programma Pymol. I modelli di L.brasilienses, e L. panamensis presentavano la migliore somiglianza, in relazione alle strutture previste secondarie e terziarie (2D-3D), presentando 15 foglie e foglie di z. Delle 15 eliche z presentate nella struttura tridimensionale del modello 8 delle eliche z erano nella stessa posizione prevista dal PSIPRED. Queste eliche corrispondono alla sequenza di amminoacidi: da 59 a 74, da 78 a 93, da 535 a 540, da 545 a 563, da 631 a 640, da 703 a 721 e da 727 a 730.
Nell’analisi generale, non vi è stata alcuna differenza esacerbata nelle quantità di foglie di z-elica e z tra le previsioni e i modelli ottenuti (tabella 3).
Tabella 3: Confronto tra le strutture secondarie previste da psipred e quella rilevata tramite Pymol.
| Modelos | α-Hélices | Folhas β | PSIPRED
α-Hélices |
PSIPRED
Folhas β |
| L.brasiliensis | 15 | 38 | 11 | 36 |
| L. donovani | 16 | 38 | 11 | 36 |
| L. infantum | 16 | 38 | 11 | 36 |
| L. mexicana | 15 | 38 | 10 | 36 |
| L. panamensis | 15 | 38 | 11 | 36 |
Fonte: Autore.
Inoltre, è stato possibile osservare che la quantità di 15 propulsori era la stessa per L.brasilienses, L. Mexicana e L. panamensis. Proprio come L.infantum e L.donovani, hanno ottenuto il numero 16 di Helix. Secondo l’albero filogenetico, questi due gruppi menzionati sono nello stesso nodo interno e sono considerati monophyletic (Figura 2).
Quindi, il RMSD (deviazione radice-media-quadrato) è stato eseguito tra i modelli e lo stampo. I valori erano promettenti, come si può vedere nella tabella 4, perché i RMSD non hanno superato il valore di 0,19. Questa scoperta può essere giustificata dall’alto grado di identità tra lo stampo e i rispettivi modelli. In generale, si prevede che le proteine con un’identità superiore al 30%, abbiano un’eccellente sovrapposizione delle catene principali, ottenendo così un RMSD dell’ordine di 2 , BENNER et al., 1997 ; CHOTHIA et al., 1986).
Tabella 4: RMSD degli ODU generati dal Modeller, avendo con orientamento gli alfa carboni dello stampo OPB di L. major.
| Molde | Modelos | RMSD (Å) |
| L. major
(2XE4) |
L.brasiliensis | 0,15 |
| L. donovani | 0,15 | |
| L. infantum | 0,16 | |
| L. Mexicana | 0,19 | |
| L. panamensis | 0,14 |
Fonte: Autore.
MAPPA DI MOLECULAR ELECTROSTATIC POTENTIAL (MEP) DELLA SURFACE OF ENZYMES AND RECEPTIVE SITES
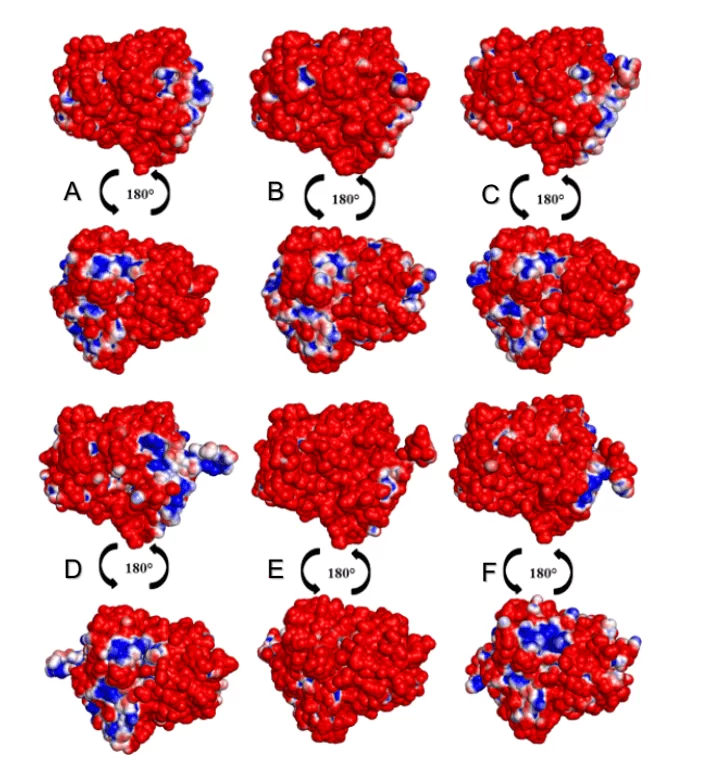
Nell’analisi degli eurodeputati delle superfici degli OBB, è stato possibile osservare che tutte le specie di leishmania presentavano una percentuale più elevata di regioni negative rispetto a quelle positive, come illustrato nella figura 15. La specie L. donovani e L. infantum (gruppo verde) ha suggerito un’area negativa (in colore blu) nella stessa regione. La specie L.brasilienses e L. panamensis (gruppo rosso) presentavano una regione negativa simile, in un modello colorimetrico simile. Entrambi i risultati possono essere giustificati dal fatto che la specie rispetto all’altro appartiene alla stessa monofila (Figura 2).
Figura 9: Mappa del potenziale elettrostatico dei modelli 3D della Leishmania spp OBB e quello del loro stampo. Para OPB, (A) L.major (molde) e modelos: (B) L. brasilienses (C) L. donovani, (D) L. infantum, (E) L. mexicana (F) L. panamensis. Nel colore blu presenta la regione positiva e nel colore rosso, la regione negativa.
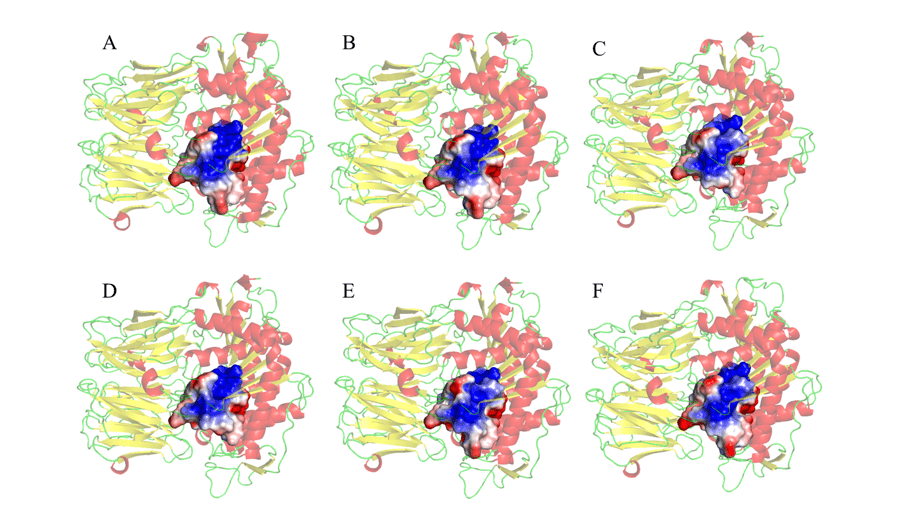
Infine, l’eurodeputato dei residui è stato eseguito anche ad un raggio di 5 – intorno alla triade catalitica, come mostrato nella Figura 10. La scelta di osservare l’eurodeputato della regione intorno alla triade catalitica è stata perché questa è la seconda regione di interazione tra l’enzima e il substrato, in cui ci sarà alloggio (ANDERSSON et al., 2010). Così, in considerazione di uno degli obiettivi dello studio, è stato possibile verificare significative somiglianze e corrispondenze tra lo stampo e i modelli testati. Questi risultati, come mostrato nella figura 10, hanno rivelato principalmente una porzione elettropositiva più grande (in blu) nella regione centrale dei siti di collegamento. Le regioni negative (in rosso) sono state osservate nelle aree periferiche dei deputati. Questi risultati sono promettenti, come si è scoperto che queste regioni sono completamente simili in tutti i modelli, e può aiutare nello sviluppo di un farmaco che ha la capacità di agire specificamente in tutti i modelli studiati.
Figura 10: Rappresentazione della potenziale mappa della superficie elettrostatica dei residui di amminoacidi, 5 o più intorno alla triade catalitica che costituiscono il sito attivo dell’enzima. (A) L.major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. In rosso sono gli elicoie z, verde le maniglie e giallo le foglie- . Nel colore dorato il sito di rilegatura. Nel colore blu presenta la regione positiva e nel colore rosso, la regione negativa.
DOGSITESCORER, CALIFORNIA
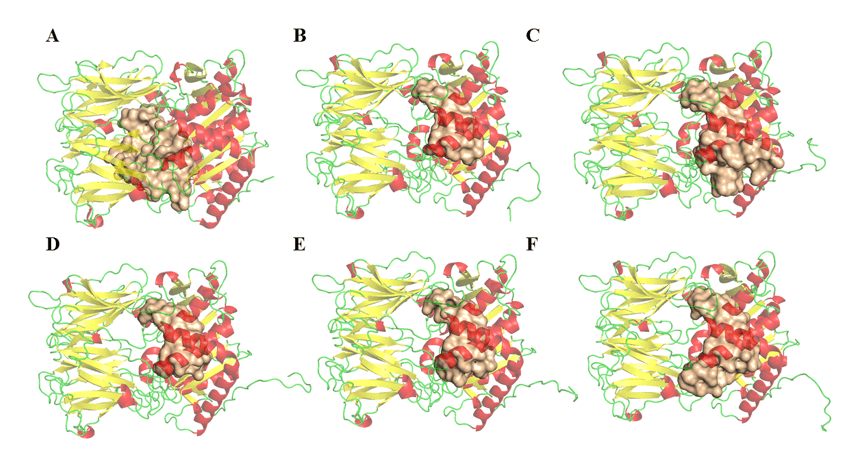
La determinazione dei parametri di volume, area e profondità dei possibili siti di legame degli enzimi opb delle specie Leishmania è stata eseguita nel programma DoGSiteScorer (http://poseview.zbh.uni-hamburg.de/) di ProteinsPlus – Structure-Based Modeling Support Server (VOLKAMER et al., 2012). Il programma indica tre siti di collegamento come descritto nella letteratura. La figura 11 mostra il confronto tra la possibile area del sito attivo e le sue differenze. Queste variabili tra gli OBB possono essere correlate a residui che non sono descritti come importanti per la loro inibizione, ma sono state considerate durante questa analisi. Pertanto, questo risultato non esclude la possibilità di antidolorio o di qualsiasi altra molecola che abbia un ampio spettro di inibizione sull’enzima, a causa del fatto che i risultati relativi al nucleo di droga erano molto simili e positivi in tutte le regioni. Tabella 5, si osserva una discrepanza relativa tra i risultati di volume e area delle cavità presenti nei modelli. Questo risultato non suggerisce una distorsione del dubbio circa il potenziale inibitorio di un singolo farmaco nei rispettivi enzimi. Tuttavia, presta attenzione al fatto che alcune proteine hanno cavità più grandi di altre. Tuttavia, queste stesse cavità hanno punti in comune, e questi sono i residui che compongono il sito catalitico, come si può vedere nella Figura 11 e possono essere esplorati.
Tabella 5: Valori che fanno riferimento alle possibili aree di connessione degli OBB (ottenuti dal server).
| Estruturas | DogSiteScoore | |||
| Volume | Área | Drug Score | ||
| OLIGOPEPTIDASE B | L. major
(PDB 2XE4) |
1690,62 | 1818,41 | 0,80 |
| L.brasiliensis | 1527,84 | 1766,63 | 0,80 | |
| L. donovani | 1074,57 | 1428,55 | 0,79 | |
| L. infantum | 1309,97 | 1572,19 | 0,80 | |
| L. mexicana | 800,92 | 799,38 | 0,85 | |
| L. panamensis | 971,96 | 1083,40 | 0,81 | |
Fonte: Autore.
Figura 11: strutture OBB e possibili aree di connessione (ottenute dal server DogSite). (A) L .major (molde)e modelos: (B) L. brasilienses, (C), L. donovani, (D) L. infantum, (E) L. mexicana e (F) L. panamensis. In rosso sono gli elicoie z, verde le maniglie e giallo le foglie- . Nel colore dorato il sito di rilegatura.
MODALITÀ NORMALE
Dopo la caratterizzazione dell’enzima nei siti strutturali, superficiali e vincolanti, le modalità normali per gli enzimi di ogni specie sono state eseguite con lo scopo dei rispettivi movimenti.
Dopo il rilassamento e il minimo di energia eseguite dalle dinamiche molecolari in GROMACS, queste strutture sono state sottoposte a normali analisi al fine di osservare possibili movimenti compatibili.
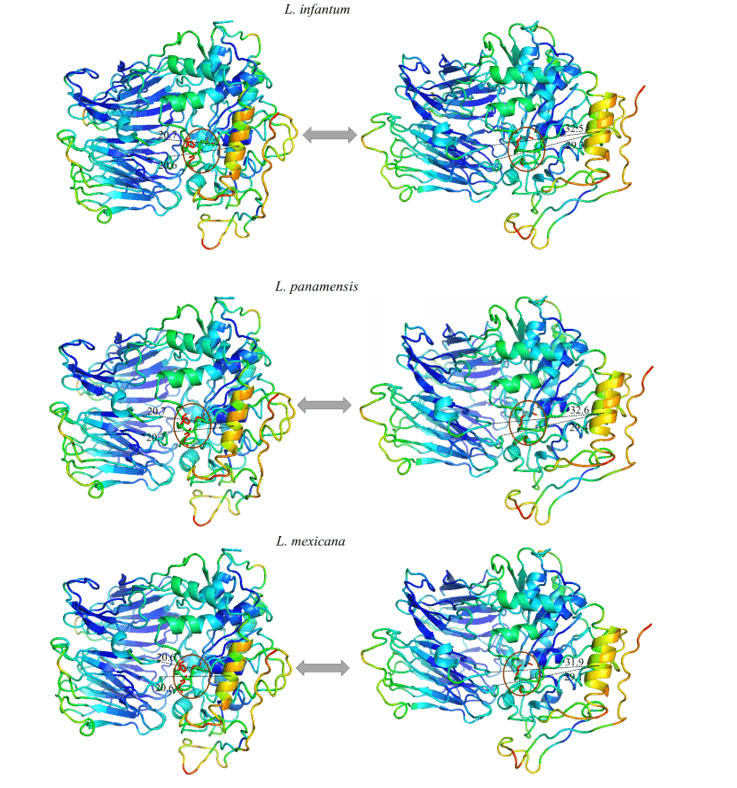
È stato possibile osservare in tutti i modelli studiato un movimento espressivo di una specifica elica, e quando si analizza questa regione nella sua composizione di amminoacidi è possibile notare che è altamente conservato. Così, il modello di movimento è stato ripetuto in tutti i modelli in questione. Questa regione suggerì un movimento lineare che si allontanava dal centro verso la periferia, esponendo la triade catalitica. Questo può essere indicativo del movimento eseguito dalla proteina per l’alloggio del substrato.
Quando si osservano le figure 12 e 13, lo stesso modello di colore può essere osservato in tutti i modelli dello studio. Questi dati rappresentano la capacità data regione deve muoversi in una certa direzione. I colori blu rappresentano regioni più invariabili durante la simulazione in base alle modalità normali, con questo, un modello di rigidità nelle proteine è evidente corrispondente principalmente alle foglie z del dominio dell’elica z e ad alcune eliche che compongono il dominio catalitico. I colori vicini al verde rappresentano le regioni intermedie in relazione alla capacità e alla gamma di movimento. Sulla base di queste informazioni, è possibile percepire questa colorazione presente in loop alle estremità dei modelli, così come in alcuni z-elicici del dominio catalitico. Infine, ci sono quelle regioni che hanno presentato una colorazione arancione/rosso, in cui è la rappresentazione di un enorme potenziale di movimento. Così, si può vedere che ci sono piccole regioni di anelli alle estremità dei domini catalitici con questo potenziale di movimento, così come una z-elica situata nella porzione del dominio catalitico, più specificamente di fronte alla triade catalitica degli OBB, in cui ha ottenuto il movimento più espressivo dello studio.
Tutti i modelli hanno ottenuto la stessa configurazione di movimento, dove un’elica a z (in arancione) si è dimostrata la regione con la più alta capacità di movimento. Pertanto, gli OBB di L. brasiliensis, L. donovani, L. infantum, L. mexicana e L. panamensis nella loro più ampia gamma di movimenti hanno ottenuto, rispettivamente, 9,8, 11,3, 11,8 , 11,9 e 11,6 gradi (figura 12 e 13).
Figura 12: Risultato dell’analisi in base alle modalità normali in rappresentazione del programma pymol, nell’immagine sono evidenziati i movimenti dei modelli, nelle loro forme rilassate (a destra) rispetto al movimento di maggiore ampiezza (a sinistra). Sono rappresentate anche le regioni più rigide (blu scuro), regioni con poco movimento (blu chiaro), regioni intermedie (verde), regioni con un buon potenziale di movimento (arancione) e regioni estremamente malleabili (rosso) (Part1/2).
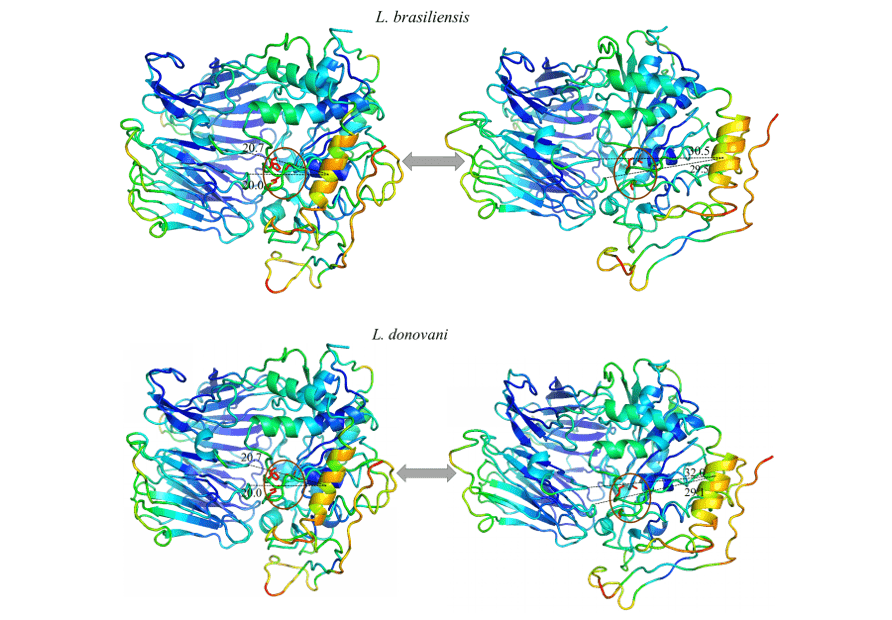
Figura 13: Risultato dell’analisi in base alle modalità normali in rappresentazione del programma pymol, nell’immagine sono evidenziati i movimenti dei modelli, nelle loro forme rilassate (a destra) rispetto al movimento di maggiore ampiezza (a sinistra). Sono rappresentate anche le regioni più rigide (blu scuro), regioni con poco movimento (blu chiaro), regioni intermedie (verde), regioni con un buon potenziale di movimento (arancione) e regioni estremamente malleabili (rosso). (Parte 2/2)
In considerazione di ciò, studi normali sostengono un possibile meccanismo di OBB non ancora descritto per le specie di Leishmania spp. fino allo studio attuale.
CONCLUSIONE
In questo studio, sono stati ottenuti i modelli tridimensionali dell’enzima opb di L.brasiliensis, L. donovani, L. infantum, L. mexicana e L. panamensis. La convalida dei modelli ha presentato risultati affidabili per tutti i modelli tridimensionali ottenuti. Nella caratterizzazione dell’enzima, la mappa del potenziale elettrostatico superficiale ha mostrato che la maggior parte dei residui presentava carica negativa. Nella caratterizzazione della regione intorno alla triade catalitica, ha dimostrato somiglianza tra volume, area e corrispondenza tra residui positivi e negativi. Pertanto, è stato possibile verificare che i risultati dell’analisi in base alle modalità normali suggerissero un movimento espressivo in una delle specifiche elica, che si verificava una distanza lineare di questo, dal centro verso la periferia, esponendo così la triade catalitica. La descrizione di questi movimenti eseguiti da questo enzima è di grande importanza per aiutare la comprensione del suo funzionamento.
Infine, i risultati del presente studio possono aggiungere conoscenze alla comunità scientifica, portando chiarimenti e nuove domande relative al tema, fungendo da base per eventuali studi nel settore sanitario.
RIFERIMENTI
A. Benner S, Cannarozzi G, Gerloff D, Turcotte M, Chelvanayagam G. Bona Fide Predictions of Protein Secondary Structure Using Transparent Analyses of Multiple Sequence Alignments. Chem Rev. 1997;97(8):2725-2844. doi:10.1021/cr940469a.
Alva, V., Nam, S. Z., Söding, J., & Lupas, A. N. (2016). The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Research, 44(W1), W410–W415. doi.org/10.1093/nar/gkw348.
Alvarenga DG, Escalda PMF, da Costa ASV, Monreal MTFD. Leishmaniose visceral: Estudo retrospectivo de fatores associados à letalidade. Rev Soc Bras Med Trop. 2010;43(2):194-197.
Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389‐3402. doi:10.1093/nar/25.17.3389.
Andersson CD, Chen BY, Linusson A. Mapping of ligand-binding cavities in proteins [published correction appears in Proteins. 2011 Apr;79(4):1363]. Proteins. 2010;78(6):1408–1422. doi:10.1002/prot.22655.
Bailey F, Mondragon-Shem K, Hotez P, et al. A new perspective on cutaneous leishmaniasis-Implications for global prevalence and burden of disease estimates. PLoS Negl Trop Dis. 2017;11(8):e0005739. Published 2017 Aug 10. doi:10.1371/journal.pntd.0005739.
Carmo RF, Luz ZMP da, Bevilacqua PD. Percepções da população e de profissionais de saúde sobre a leishmaniose visceral. Cien Saude Colet. 2016;21(2):621-628. doi:10.1590/1413-81232015212.10422015.
Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5(4):823‐826.
Derewenda ZS, Derewenda U, Kobos PM. (His)Cε-H···O=C< Hydrogen Bond in the Active Sites of Serine Hydrolases. J Mol Biol. 1994;241(1):83-93. doi:10.1006/JMBI.1994.1475.
Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32(Web Server issue):W665–W667. doi:10.1093/nar/gkh381.
Eisenberg, D., Lüthy, R., & Bowie, J. U. (1997). [20]VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods in Enzymology, 277, 396–404. https://doi.org/10.1016/S0076-6879(97)77022-8.
Eyal E, Lum G, Bahar I. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics. 2015;31(9):1487–1489. doi:10.1093/bioinformatics/btu847.
Ghorbani Masoud, Farhoudi Ramin. Leishmaniasis in humans: drug or vaccine therapy? Drug Des Devel Ther. 2018;12:25-40. doi:10.2147/DDDT.S146521.
Hedstrom L. Serine Protease Mechanism and Specificity. Chem Rev. 2002;102(12):4501-4524. doi:10.1021/cr000033x.
Katsila T, Spyroulias GA, Patrinos GP, Matsoukas MT. Computational approaches in target identification and drug discovery. Comput Struct Biotechnol J. 2016;14:177–184. Published 2016 May 7. doi:10.1016/j.csbj.2016.04.004.
Kumar S, Tamura K, Nei M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5(2):150-163.
Macedo-Silva RM, dos Santos C de LP, Diniz VA, De Carvalho JJ, Guerra C, Côrte-Real S. Peripheral blood fibrocytes: New information to explain the dynamics of Leishmania infection. Mem Inst Oswaldo Cruz. 2014;109(1):61-69. doi:10.1590/0074-0276130247
Machado P de A, Carneiro MPD, Sousa-Batista A de J, et al. Leishmanicidal therapy targeted to parasite proteases. Life Sci. 2019;219:163-181. doi:10.1016/J.LFS.2019.01.015.
Morris, A. L., MacArthur, M. W., Hutchinson, E. G., & Thornton, J. M. (1992). Stereochemical quality of protein structure coordinates. Proteins: Structure, Function, and Bioinformatics, 12(4), 345–364. https://doi.org/10.1002/prot.340120407.
Ovchinnikova M V., Mikhailova AG, Karlinsky DM, Gorlenko VA, Rumsh LD. Reversible cyclic thermal inactivation of oligopeptidase B from Serratia proteamaculans. Acta Naturae. 2018;10(2):65-70.
Ramachandran, G. N., Ramakrishnan, C., & Sasisekharan, V. (1963). Stereochemistry of polypeptide chain configurations. Journal of Molecular Biology, 7(1), 95–99. https://doi.org/10.1016/S0022-2836(63)80023-6.
Santos Filho, O. A., & Alencastro, R. B. de. (2003). Modelagem de proteínas por homologia. Química Nova, 26(2), 253–259. https://doi.org/10.1590/S0100-40422003000200019
Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. Published 2011 Oct 11. doi:10.1038/msb.2011.75.
SODERO ACR, DOS SANTOS ACGO, MELLO JFRE, et al. Oligopeptidase B and B2: comparative modelling and virtual screening as searching tools for new antileishmanial compounds. Parasitology. 2017;144(4):536-545. doi:10.1017/s0031182016002237.
Swenerton RK, Zhang S, Sajid M, et al. The oligopeptidase B of Leishmania regulates parasite enolase and immune evasion. J Biol Chem. 2011;286(1):429-440. doi:10.1074/jbc.M110.138313.
Volkamer, A., Kuhn, D., Rippmann, F., & Rarey, M. (2012). Dogsitescorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics, 28(15), 2074–2075. https://doi.org/10.1093/bioinformatics/bts310.
Wang Q, Arighi CN, King BL, et al. Community annotation and bioinformatics workforce development in concert–Little Skate Genome Annotation Workshops and Jamborees. Database (Oxford). 2012;2012:bar064. Published 2012 Mar 20. doi:10.1093/database/bar064
Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics. 2016;54:5.6.1–5.6.37. Published 2016 Jun 20. doi:10.1002/cpbi.3.
Wiederstein, M., & Sippl, M. J. (2007). ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research, 35(SUPPL.2), 407–410. https://doi.org/10.1093/nar/gkm290.
WHO. Integrating Neglected Tropical Diseases into Global Health and Development: Fourth WHO Report on Neglected Tropical Diseases.; 2017. http://apps.who.int/iris/bitstream/10665/255011/1/9789241565448-eng.pdf?ua=1.
WHO. (2019). Leishmanioses – Informe Epidemiológico das Américas No 7 – Março, 2019. Retrieved from http://iris.paho.org/xmlui/bitstream/handle/123456789/50505/ 2019-cde-leish-informe-epi-das-americas.pdf?sequence=2&isAllowed=y.
ANNEX – FIGURES E TABLES IN ITALIANO
Schema 1: schema semplificato di passaggi e metodi del materiale.
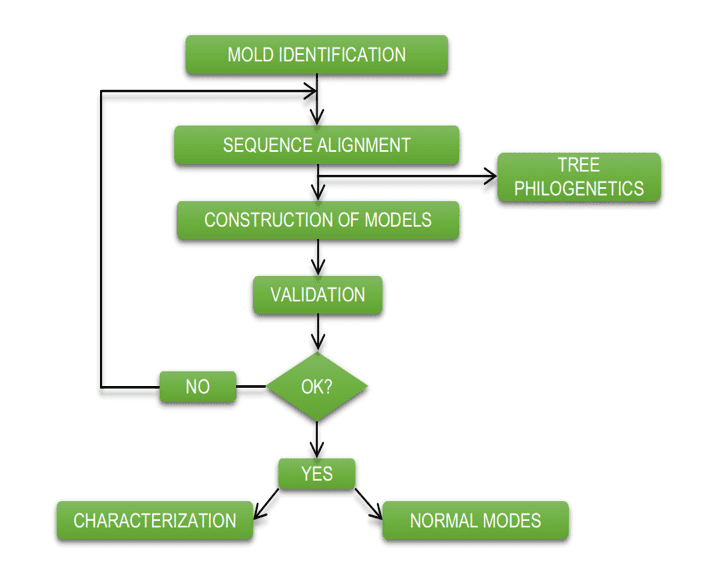
Tabella 1: Percentuale di identità tra i modelli di oligopeptity B delle specie di Leishmania e la loro rispettiva muffa.
| Mold protein (code PDB) | Models OPB
(código uniprot) |
Identity (%) | Gaps (%) |
| OPB
L. major (code PDB 2XE4) |
L.brasiliensis
(A4H5Q8) |
86 | 0 |
| L. donovani
(C9EF60) |
96 | 0 | |
| L. infantum
(A4HTZ8) |
96 | 0 | |
| L. Mexicana
(E9AMS8) |
90 | 0 | |
| L. panamensis
(A0A088RJA7) |
86 | 0 |
Fonte: Autoral.
Figura 6: Risultati dei grafici ramachandran, ottenuti dal programma PROCHECK, dalle strutture dei modelli OBB generati e dallo stampo.
| Structures | % waste in the regions | ||||
| Favorable | Allowed | Unfavorable | |||
| OLIGOPEPTIDASE B | L. major (A)
(PDB 2XE4) |
90,2 | 9,5 | 0,3 | |
| L.brasilienses (B) | 92,2 | 7,7 | 0,2 | ||
| L. donovani (C) | 91,9 | 8,0 | 0,2 | ||
| L. infantum (D) | 92,3 | 7,3 | 0,3 | ||
| L. Mexicana (E) | 91,2 | 8,3 | 0,5 | ||
| L. panamensis (F) | 91,4 | 8,3 | 0,3 | ||
Fonte: Preparato dall’autore in base ai risultati di procheck.
Figura 7: Risultati del punteggio z calcolati sul server ProSA-web delle strutture di stampo (per confronto). (A) L .major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. La regione in blu scuro indica il punteggio delle proteine ottenute dalla NMR e in azzurro delle proteine ottenute dalla diffrazione a raggi X
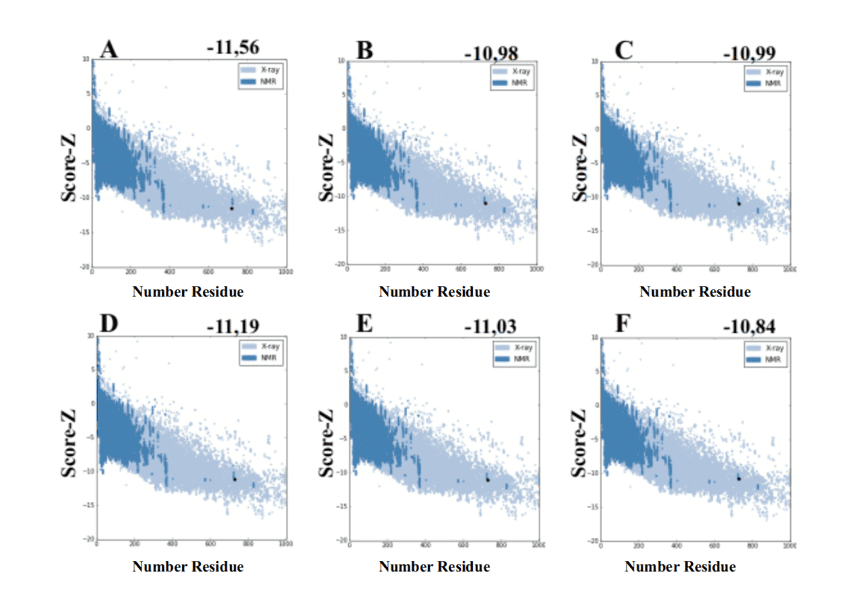
Tabella 2: Risultati di Verifica 3D, che mostra la percentuale di residui con punteggio > 0.2.
| Structures | % residue with score > 0,2 | |
| OLIGOPEPTIDASE B | L.major
(PDB 2XE4) |
93,20 |
| L.brasilienses | 95,62 | |
| L.donovani | 97,12 | |
| L.infantum | 94,93 | |
| L.mexicana | 95,62 | |
| L.panamensis | 94,93 |
Fonte: Autore.
Tabella 3: Confronto tra le strutture secondarie previste da psipred e quella rilevata tramite Pymol.
| Models | α-Hélix | Sheet β | PSIPRED
α-Hélix |
PSIPRED
Sheet β |
| L.brasiliensis | 15 | 38 | 11 | 36 |
| L. donovani | 16 | 38 | 11 | 36 |
| L. infantum | 16 | 38 | 11 | 36 |
| L. mexicana | 15 | 38 | 10 | 36 |
| L. panamensis | 15 | 38 | 11 | 36 |
Fonte: Autore.
Tabella 4: RMSD degli ODU generati dal Modeller, avendo con orientamento gli alfa carboni dello stampo OPB di L. major.
| Mold | Models | RMSD (Å) |
| L. major
(2XE4) |
L.brasiliensis | 0,15 |
| L. donovani | 0,15 | |
| L. infantum | 0,16 | |
| L. mexicana | 0,19 | |
| L. panamensis | 0,14 |
Fonte: Autore.
Tabella 5: Valori che fanno riferimento alle possibili aree di connessione degli OBB (ottenuti dal server).
| Structures | DogSiteScoore | |||
| Volume | Area | Drug Score | ||
| OP OLIGOPEPTIDASE B | L. major
(PDB 2XE4) |
1690,62 | 1818,41 | 0,80 |
| L.brasiliensis | 1527,84 | 1766,63 | 0,80 | |
| L. donovani | 1074,57 | 1428,55 | 0,79 | |
| L. infantum | 1309,97 | 1572,19 | 0,80 | |
| L. mexicana | 800,92 | 799,38 | 0,85 | |
| L. panamensis | 971,96 | 1083,40 | 0,81 | |
Fonte: Autore.
[1] Laurea in Biochimica Medica e Master.
[2] Master in Scienze Farmaceutiche e Farmaceutiche presso UFRJ.
[3] Dottorato di ricerca in Chimica, Master in Chimica Organica e Prodotti Farmaceutici Industriali.
Inviato: Maggio, 2020.
Approvato: maggio 2020.