ORIGINAL-ARTIKEL
RIBEIRO, Fernando de Sá [1], JESUS, Jéssica Barbosa de [2], SOUZA, Alessandra Mendonça Teles de [3]
RIBEIRO, Fernando de Sá. JESUS, Jéssica Barbosa de. SOUZA, Alessandra Mendonça Teles de. Analyse und Charakterisierung eines vielversprechenden therapeutischen Ziels, das in Leishmania spp identifiziert wurde. Revista Científica Multidisciplinar Núcleo do Conhecimento. Ano 05, Ed. 05, Vol. 09, pp. 99-132. Maio de 2020. ISSN: 2448-0959, Zugangslink: https://www.nucleodoconhecimento.com.br/gesundheit/zieltherapeutische
ZUSAMMENFASSUNG
Leishmaniose ist eine vernachlässigte Krankheit, die durch Protozoen der Gattung Leishmania spp. verursacht wird, von der jedes Jahr etwa 1,6 Millionen Menschen betroffen sind und 500.000 sich in der viszeralen Form präsentieren. In Brasilien gibt es jährlich etwa 30.000 neue Fälle. Darüber hinaus ist das Land für 90% der gemeldeten Fälle von Viszeraler Leishmaniose verantwortlich, und dies ist eine schwerere Form der Krankheit. In Verbindung mit diesen Tatsachen ist die derzeitige Behandlung wirkungslos und trägt zur Etablierung resistenter Stämme bei. Derzeit hat die Behandlung mehrere Nebenwirkungen und bleibende Schäden für die Gesundheit der Patienten, diese Tatsache hat zur Suche nach neuen Medikamenten gegen Leishmaniose beigetragen. Das Enzym Oligopeptidase B (OPB) wurde als mögliches therapeutisches Ziel bei der Entwicklung von antiparasitären Wirkstoffen untersucht. Ziel dieser Arbeit ist es daher, das dreidimensionale Modell des Enzyms Oligopeptidase B verschiedener Arten von Leishmania spp zu konstruieren. und vergleichen Sie sie miteinander. Zu diesem Zweck wurde die vergleichende Modellierungsmethode verwendet. Bei dieser Methode wurden die Modelle der Arten L. brasiliensis, L. donovani, L. infantum, L. mexicana und L. panamensis mit dem MODELLER-Programm konstruiert. Sobald die Modelle fertig waren, wurde der Validierungsprozess durchgeführt und anschließend charakterisiert, was eine vielversprechende Ähnlichkeit zwischen den Modellen überprüfen konnte. Schließlich wurden diese Modelle der Analysemethode durch normale Modi unterzogen, die ein ähnliches Bewegungsmuster erhielten, so dass es möglich war, eine Bewegung in einem bestimmten Bereich einer Alpha-Helix zu überprüfen, was zu der Triade des Enzyms führte, die exponiert wurde, was auf einen Wirkmechanismus hindeuten kann. Schließlich wird erwartet, dass sie die Modelle verwendet, die gebaut wurden, um bei der Entwicklung einer vielversprechenden neuen Therapie zur Behandlung von Leishmaniose zu helfen.
Schlüsselwörter: Leishmaniose, Oligopeptidase B, molekulare Modellierung, normale Modi.
EINFÜHRUNG
Leishmaniose, verursacht durch Leishmanie spp., ist eine Krankheit, die durch verschiedene Arten von Manifestationen gekennzeichnet ist, von milden, in denen es Berichte über kleine Läsionen gibt, die auch ohne ordnungsgemäße Behandlung auf die schwersten wie Viszerale Leishmaniose (VL) zurückgehen. In Brasilien, die schwerste Form dieser Krankheit präsentiert alarmierende Daten im Vergleich zu anderen Ländern, so dass das Land der größte Inhaber von VL-Fälle in ganz Amerika (ALVARENGA, 2010; WER, 2019).
Diese Krankheit gehört zu der Gruppe der vernachlässigten Krankheiten, die zu all jenen Krankheiten gehören, die hauptsächlich unterentwickelte Länder und ärmere Regionen betreffen, so dass sie kein Interesse an der Entwicklung von Arzneimitteln weckt. Daher ist es notwendig, dass effiziente und kostengünstige Techniken überleben, um diesen Mangel an finanziellen Anreizen für die Untersuchung dieser Krankheit zu überwinden. So können Rechenmethoden eingesetzt werden, um die Zeit bei der Entwicklung einer vielversprechenden neuen Therapie und damit die Kosten im Vergleich zu traditionelleren Methoden zur Arzneimittelentwicklung zu reduzieren (BAILEY et al., 2017; WER, 2017).
Trotz der geringen Investitionen in diesem Bereich, gibt es Behandlung für die Krankheit wie die antimonialen Pentas valentes (Medikament erster Wahl) oder Amphotericin B (Medikament der zweiten Wahl). Solche Behandlungen haben jedoch mehrere Nachteile wie die hohe Resistenzrate und die Vielzahl von Nebenwirkungen, die von Seekrankheit über mögliche Probleme bis hin zum dritten (3.) Paar des Hirnnervs reichen, was zu motorischen Schwierigkeiten führt (MACEDO-SILVA et al., 2014).
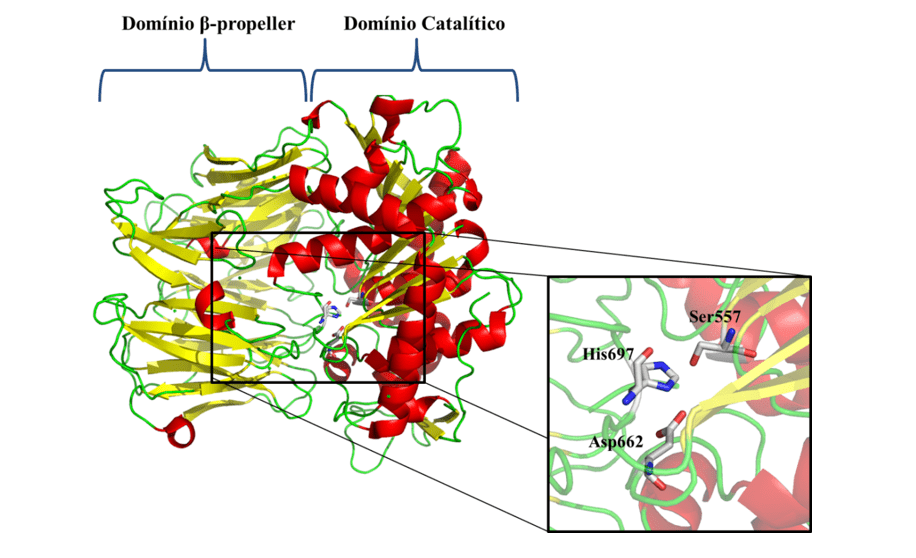
Angesichts der auftretenden Probleme und der epidemiologischen Bedeutung der Leishmaniose ist die Notwendigkeit der Untersuchung neuer spezifischer therapeutischer Ziele gegen die Krankheit berüchtigt. Als eines der neuen therapeutischen Ziele haben wir Oligopeptidase B (OPB) (Abbildung 1), eine Serinprotease, die zur Unterfamilie S9A gehört und als charakteristisch eine katalytische Triade hat, die aus den Rückständen von Aminosäuren Serin (Ser), Aspinsäure (Asp) und Histidin (Sein) besteht, die sich zwischen den beiden Domänen befinden. Dieses Enzym hat als charakteristisch die Fähigkeit, Rückstände von Proteinstrukturen zu spalten. Darüber hinaus wird es in der Literatur als schlüsselfertiger Bestandteil für den Immun-Fluchtmechanismus des Parasiten beschrieben, das die Enolase, ein Protein, das das Protozoen opsonisar, so dass, wenn es durch das Makrophagen erkannt wird, wird es zerstört. Während sich der Parasit jedoch innerhalb des Makrophagens befindet, ist das OPB super exprimiert, was dazu führt, dass der Parasit in der Zelle nicht erkannt wird, so dass er sich vermehrt, bis er geglättet ist (SODERO et al., 2016; SWENERTON et al., 2011; OVCHINNIKOVA et al. 2018).
Abbildung 1: Dreidimensionale Struktur der L. Major OPB (PDB-Code 2XE4) (MCLUSKEY et al., 2010) mit den katalytischen und Sekundäre Strukturen wie z.B. ‘-helix,’-Blätter und Griffe sind rot, gelb und grün dargestellt. Insbesondere werden die Rückstände (Ser557, Asp662 und His697) der katalytischen Triade gezeigt.
Daher zielte die fragliche Studie darauf ab, die OPBs und ihre aktiven Standorte der Arten L. brasiliensis, L, donovani, L. infantum, L. mexicana und L. panamensis zu untersuchen und zu charakterisieren. Schließlich wurde erwartet, dass mögliche Ähnlichkeiten zwischen Proteinen identifiziert werden, so dass es die zukünftige Entwicklung einer vielversprechenden neuen Therapie mit einem breiten Wirkungsspektrum auf Enzyme aller Arten in der Studie ermöglichen würde.
ALLGEMEINES ZIEL
Angesichts der Notwendigkeit der Entwicklung neuer chemischer Einheiten für die Behandlung gegen Leishmaniose hatte diese Arbeit als Hauptziel den Aufbau und die Charakterisierung des Enzyms Oligopeptidase B von Leishmania spp. mit Hilfe von Computerstudientechniken.
SPEZIFISCHE ZIELE
- Konstruieren und validieren sie die Modelle von Oligopeptidase-B-Enzymen von Leishmanien-Arten;
- Führen Sie die Charakterisierung von Enzymen durch;
- Führen Sie Simulationsstudien nach normalen Modi durch.
MATERIAL UND METHODEN
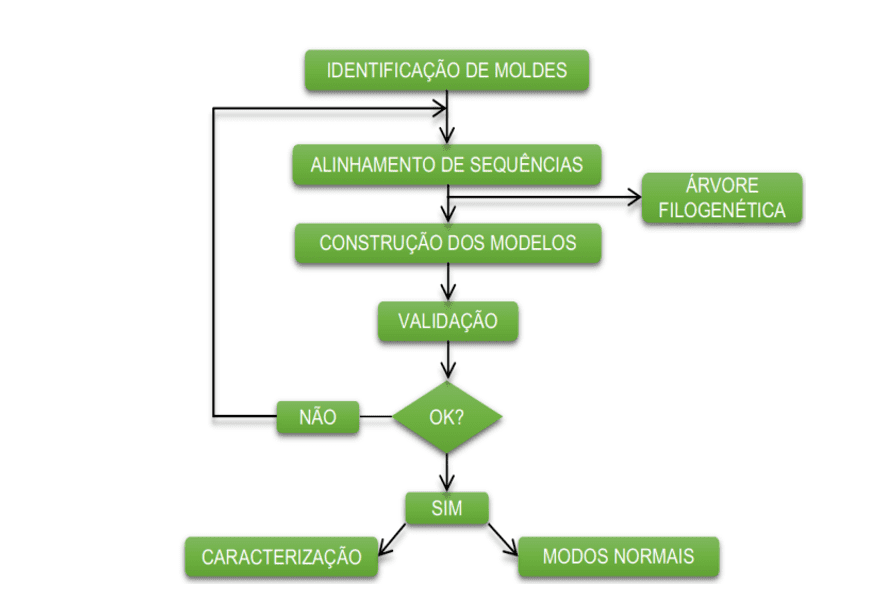
Das folgende Diagramm (Abbildung 1) zeigt vereinfacht die Schritte, die bei der Entwicklung dieser Arbeit verwendet werden.
Schema 1: Vereinfachtes Schema der Materialschritte und -methoden.
ERHALTEN DER PRIMÄREN STRUKTUR
Die primären Strukturen von Leishmanien OPBs wurden aus der Uniprot-Datenbank (The Universal Protein Resource) (WANG et al., 2012) gewonnen.
MOLD ID
Anschließend erfolgte die Suche nach Schimmelpilzproteinen aus der Ausrichtung zwischen der Aminosäuresequenz des Zielproteins und den Aminosäuresequenzen von Proteinen, die vom BLAST-Server (Basic Local Alignment Search Tool) (ATLSCHUL et al., 1997) auf der Grundlage der Identität zwischen Aminosäuresequenzen in der Proteindatenbank (PDB) abgelagert wurden.
ALIGNMENT VON SEQUENCES
Sobald die zu verwendende Form definiert war, wurde die Formfolge im ClustalOmega-Programm (ClustaO) (SIEVERS et al., 2011) an ihren jeweiligen OPBs ausgerichtet.
MODELLBAU
Nach Erhalt der Ausrichtung wurden diese Informationen im ModellER v9.20-Programm verwendet, um die 3D-Strukturen der Modelle zu konstruieren (WEBB und SALI, 2016).
VALIDIERUNG VON GEBAUTEN MODELLEN
Für die Validierung der Modelle wurde das Ramachandran-Diagramm verwendet, das vom PDBsum-, Verify-3D- und ProSA-Webserver generiert wurde und alle vom Saves-Server abgerufen wurden.
Bei der Analyse des Ramachandran-Graphen ist es zulässig, die Dihedralwinkel (phi () und psi () von Aminosäurerückständen in der Proteinstruktur zu visualisieren. Der phi-Winkel () ist das Ergebnis der Verbindung zwischen der NH-Gruppe und Alpha-Kohlenstoff, während der psi-Winkel () aus der Verbindung zwischen Alpha-Kohlenstoff und der Carbonylgruppe stammt (RAMACHANDRAN et al., 1963).
Das Diagramm bietet eine einfache Möglichkeit, die Verteilung der Tormwinkel einer Proteinstruktur zu visualisieren. Darüber hinaus bietet es einen Überblick über die zulässigen und nicht zulässigen Regionen der Tormwinkelwerte und dient als wichtiger Faktor bei der Beurteilung der Qualität von 3D-Proteinstrukturen. Damit definiert er die Rückstände, die in den energetisch günstigeren und ungünstigeren Regionen zu finden sind, und leitet die Bewertung der Qualität theoretischer oder experimenteller Proteinmodelle. Diese Grafik ist in Regionen unterteilt, so dass die günstigsten Regionen in rot sind, zusätzliche Regionen, die in braunen, freizügigen Regionen in gelben und nicht-zulässigen Regionen in Weiß freizügiger sind. Nach dieser Validierung für ein vorhergesagtes Modell, das von ausgezeichneter Qualität betrachtet werden soll, muss es mehr als 90% der Aminosäurerückstände in der günstigsten Region aufweisen (SANTOS-FILHO und ALENCASTRO, 2003).
Darüber hinaus muss es die Mehrheit seiner Rückstände in den günstigsten Regionen haben, sowie keine Rückstände in den nicht-zulässigen Regionen, mit Ausnahme der Aminosäuren Glycin (Gly) und Prolin (Pro), die Ausnahmen für diesen Bereich sind. Diese beiden Rückstände stellen Variationen in der seitenkette dar, die im Fall von Pro eine größere Steifigkeit verleihen, und eine größere Flexibilität, im Fall von Gly, so dass das Sein unerwartete Winkelannehmen annehmen kann. Aus diesem Grund werden sie in den nicht zulässigen Regionen des Diagramms akzeptiert. Die Existenz der nicht zugelassenen Regionen ist darauf zurückzuführen, dass es unter den Rückständen (Seitenketten) von Aminosäuren sterische Wirkungen gibt. (MORRIS et al., 1992).
Verify 3D analysiert die Kompatibilität eines atomaren (3D) Modells mit einer eigenen Aminosäuresequenz (1D). Jeder Rückstand erhält eine Strukturklasse basierend auf seiner Lage und Umgebung (Alpha, Beta, Schleife, Polar, Apolar, etc.). Bald darauf wird eine Datenbank verwendet, die aus guten Strukturen generiert wird, um eine Punktzahl für jede der 20 Aminosäuren in dieser Strukturklasse zu erhalten. Die vertikale Achse im Diagramm stellt den Mittelwert des 3D-1D-Profils für jedes Residuum in einem Schiebefenster von 21 Residuen dar, und die Ergebnisse in Form von Partituren reichen von -1 (schlechter Punktzahl) bis +1 (gute Punktzahl) (EISENBERG et al., 1997).
ProSa-web berechnet eine Gesamtqualitätsbewertung für eine bestimmte Eingabestruktur. Wenn die Punktzahl außerhalb eines Bereichs ist, der für native Proteine charakteristisch ist, enthält die Struktur wahrscheinlich Fehler. Viele lokale Qualitätsbewertungen deuten auf problematische Teile des Modells hin, die auch in einem 3D-Molekülbetrachter zur einfachen Erkennung hervorgehoben werden (WIEDERSTEIN und SIPPL, 2007).
ANALYSE DER SEKUNDÄRSTRUKTUR
Nach der Validierungsphase wurden die tertiären Strukturen der Modelle mit der Verteilung der sekundären Struktur verglichen, die von Quick2D vorhergesagt wurde und auf dem Bioinformatics Toolkit Server (https://toolkit.tuebingen.mpg.de/ verfügbar ist) und somit die Modelle ausgewählt wurden (ALVA et al., 2016).
CHARAKTERISIERUNG VON OLIGOPEPTIDASES (OPBS) VON LEISHMANIAS
ELEKTROSTATISCHE POTENTIALKARTE (MEP)
Um die MdEP der Oberflächen und der Verbindungsstellen der OPBs zu erhalten, wurde eine Erweiterung des PyMOL-Programms, apb tools (BAKER et al., 2001) verwendet. Vor der Analyse in PyMOL wurden die Enzyme im PDB2PQR Server (http://nbcr-222.ucsd.edu/pdb2pqr_2.0.0/) unter Verwendung des Standard-Serverparameters (DOLINSKY et al., 2004) hergestellt.
CHARAKTERISIERUNG VON OPBS-SITES UND SUBSITES
Die Modelle und die Form wurden in der Proteine plus Plattform mit der Dogsitescorer-Option (VOLKAMER et al., 2012) eingereicht, in der Hohlräume in den 3D-Strukturen der Modelle vorhergesagt wurden. Daraus wurden Ergebnisse zu möglichen Bindungsstellen und Enzymuntersiten zur Vorhersage dieser Hohlräume generiert. Das Programm verwendet ein dreidimensionales Netz, dessen Kante zwischen 0,2 und 1,0 ° eingestellt werden kann, zusammen mit einem Gaußschen Filter, der verwendet wird, um Hohlräume auf der Oberfläche des Proteins zu identifizieren, die für die Aufnahme von Ligandenatomen geeignet sind. Darüber hinaus sagt DoGSiteScorer auch die Arzneimittelfähigkeit für jeden vorhergesagten Hohlraum voraus. So wird für jede Wechselwirkung zwischen Protein und dem möglichen Medikament eine Punktzahl zugewiesen, die sich auf die Kavitätsmedikamentität bezieht, die als Drugscore bezeichnet wird. Um den Wert von drugscore vorherzusagen, verwendet das Programm eine Support-Vektormaschine (SVM), bei der die folgenden Deskriptoren verwendet werden: Volumen, Anteil unpolarer Rückstände und Tiefe (VOLKAMER et al., 2012).
PHYLOGENETIC TREE
Schließlich wurde der Grad der evolutionären Verwandtschaft zwischen Leishmanen-Arten mit dem MEGA-Programm (Molecular evulutionary genetics analysis) analysiert, mit hilfe der Neighbor-Joining-Methoden, die den Aufbau des phylogenetischen Baumes ermöglichen, um die evolutionären Proximitäten zwischen Populationen von Sequenzen zu definieren, die zuvor vom Benutzer definiert wurden (KUMAR et al., 2004).
NORMAL MODES
Um die normalen Modi auszuführen, wurden die Dateien verwendet, die in den Stadien der Energie-Minimiza-Es von GROMACs Version 5.1.2 erzeugt wurden. Die ersten 4 Schritte im Zusammenhang mit dem molekularen Dynamikprozess wurden durchgeführt. Die erste war die Generierung von Topologie-Dateien, mit der Zugabe von Wasserstoff endematosien. In der zweiten wurde die Wasserbox erstellt, und dies ist ein sehr wichtiger Schritt für die Berechnung der Wechselwirkung zwischen dem Protein und dem Lösungsmittel. In der dritten wurden Ionen hinzugefügt, um ein neutrales System zu etablieren. Schließlich wurden in der vierten Stufe Energieminimierungen durchgeführt, in die das Forcefeld AMBER99SB eingefügt wurde. Von diesem Zeitpunkt an wurden die normalen Modi der Modelle mit dem ANM (Anisotropic Network Model) (http://anm.csb.pitt.edu/) Server durchgeführt, um die Bewegung von Enzymen von Leishmanienarten zu analysieren und auch einige wichtige Eigenschaften für Enzyme zu beobachten, wie mögliche Bewegungen im Zusammenhang mit dem Wirkmechanismus (EYAL et al., 2015).
ERGEBNISSE UND DISKUSSIONEN
SELECTION VON MOLD PROTEINS UND ALIGNMENT BETWEEN SEQUENCES
Wir erhielten 100 primäre Strukturen der OPBs der Leishmanie spp. mit dem UniProt-Server, wobei 5 Arten von Leishmanien ausgewählt wurden. Die überarbeiteten Aminosäuresequenzen, die aus dem OPB-Enzym ausgewählt wurden, waren L.brasiliensis, L. donovani, L. infantum, L. mexicana und L. panamensis unter den Codes A4H5Q8, C9EF60, A4HTZ8, E9AMS8 und A0A088RJA7, bzw. Diese Arten wurden aufgrund ihrer hohen Inzidenz in Südamerika und ihrer Resistenz gegen die aktuelle Behandlung von Leishmaniose (GHORBANI und FARHOUDI, 2018) ausgewählt.
Für die Konstruktion von 3D-Modellen wurde das BLAST-Programm verwendet, um die Aminosäuresequenzen der Zielsequenzen mit Proteinsequenzen experimentell aufgeklärter dreidimensionaler Strukturen zu vergleichen. Basierend auf der Identität zwischen den Sequenzen und der Anzahl der Lücken wurde die ENZYME OPB von L. major (Code PDB 2XE4) als Schimmelprotein ausgewählt (MCLUSKEY et al., 2010). Die Identität zwischen den Zielenzymen und ihrer jeweiligen Form zeigte Werte zwischen 86% und 96% (Tabelle 1). Der Prozentsatz der Identität zwischen zwei Sequenzen bezieht sich auf das Vorhandensein derselben Aminosäure in der gleichen Position zwischen den ausgerichteten Sequenzen. Für die Konstruktion eines Modells eines Proteins mit mehr als 80 Aminosäurerückständen sollte der Identitätsanteil zwischen den primären Strukturen der Form und dem Modell über 25 % liegen. Darüber hinaus muss der Prozentsatz der Lücken niedrig sein von 20 %, um als eine gute Ausrichtung angesehen werden zu können (SANTOS-FILHO und ALENCASTRO, 2003). Somit ist die Wahrscheinlichkeit der Ähnlichkeit der dreidimensionalen Strukturen von Proteinen hoch.
Tabelle 1: Prozentsatz der Identität zwischen den Modellen der Oligopeptität B der Leishmanischen Art und ihrer jeweiligen Form.
| Proteína molde (código PDB) | Modelos de OPB
(código uniprot) |
Identidade (%) | Gaps (%) |
| OPB
L. major (código PDB 2XE4) |
L.brasiliensis
(A4H5Q8) |
86 | 0 |
| L. donovani
(C9EF60) |
96 | 0 | |
| L. infantum
(A4HTZ8) |
96 | 0 | |
| L. Mexicana
(E9AMS8) |
90 | 0 | |
| L. panamensis
(A0A088RJA7) |
86 | 0 |
Quelle: Authoral.
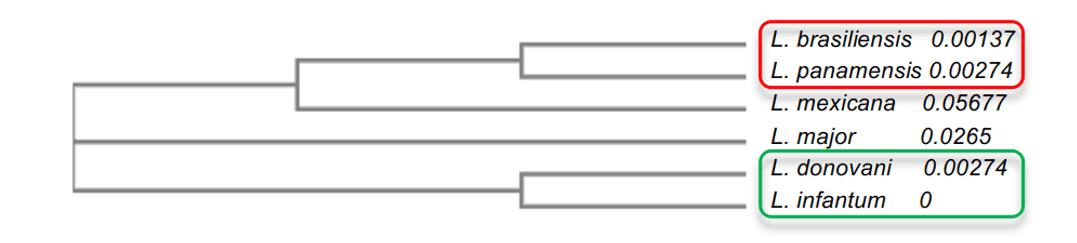
In der phylogenetischen Baumanalyse konnte der Identitätsgrad der Leishmanie-Arten erklärt werden. Es wurde beobachtet, dass die Arten mit höherem Identitätsgrad, wie L. infantum und L. donovani, beide mit 96%, eine Nähe zueinander darstellten, abgesehen davon, dass sie näher an L.major waren. Die anderen Arten präsentierten niedrigere Werte wie L.mexicana (90%), die im Median im Vergleich zu den anderen Modellen war. Schließlich waren die Modelle mit dem niedrigsten Identitätsanteil, L. brasiliensis und L. panamensis, beide mit 86%, evolutionär weiter von ihrer Form entfernt, aber sehr nahe beieinander (Abbildung 2). Diese Analyse ermöglichte das Verständnis des Unterschieds zwischen dem Grad der Identität zwischen den Arten und dem Verständnis einiger wichtiger Merkmale des Enzyms zwischen den Arten.
Abbildung 2: Phylogenetisches Baumschema von Leishmania spp. OPBs Enzymen. In dieser Darstellung werden die Gruppen mit der höchsten Ähnlichkeit zueinander dargestellt, wobei Gruppe 1 rot und Gruppe 2 grün hervorgehoben ist.
DREIDIMENSIONALE MODELLE DES ENZYMS DER LEISHMANISCHEN ARTEN
Die 3D-Modelle der Art wurden mit dem Modeller-Programm (WEBB und SALI, 2016) aus der Ausrichtung der Primärstrukturen konstruiert (Abbildung 3 und 4).
Abbildung 3: Ergebnis der Ausrichtung zwischen den primären Sequenzen der in UNIPROT ausgewählten OPBs (Teil 1/2).

Abbildung 4: Ergebnis der Ausrichtung zwischen den primären Sequenzen der in UNIPROT ausgewählten OPBs (Teil 2/2).

Bei der Beobachtung des Ergebnisses der Ausrichtung der Strukturen (Abbildungen 3 und 4) wurde ein höherer Grad an Ähnlichkeit zwischen L. donovani und L. infantum (Gruppe 2) sowie eine größere Übereinstimmung zwischen L. brasiliensis und L. panamensis (Gruppe 1) beobachtet. Diese Ergebnisse bestätigen, was der phylogenetische Baum in Abbildung 2 darstellt, wo eine Nähe zwischen den Arten L. donovani und L. infantum und eine weitere Nähe zwischen den Arten von L. brasiliensis und L. panamensis suggeriert wird. Auf der anderen Seite stellten die beiden genannten Gruppen eine größere evolutionäre Distanz im Vergleich zueinander dar. Diese Tatsache ist berüchtigt, wenn ein Vergleich in der Ausrichtungsanalyse gemacht wird, bei der die primären Strukturen einen größeren Unterschied zwischen den Rückständen im Vergleich zu den beiden Gruppen zeigten. Darüber hinaus erwies sich das Ergebnis der Angleichung von L. mexicana als sehr vielversprechend, was sich auf die Unterstützung dieser vorgeschlagenen Diskussion bezieht, da die im Vergleich zu den anderen Arten beobachteten Unterschiede gleichwertig waren, manchmal der ersten genannten Gruppe, manchmal der zweiten. In einigen Teilen der analysierten Sequenz wurden auch spezifische Mutationen beobachtet, die nur für L. mexicana charakteristisch sind. Somit kann diese Tatsache aufgrund ihrer Position im Median im phylogenetischen Baum im Verhältnis zu den anderen Arten bestätigt werden.
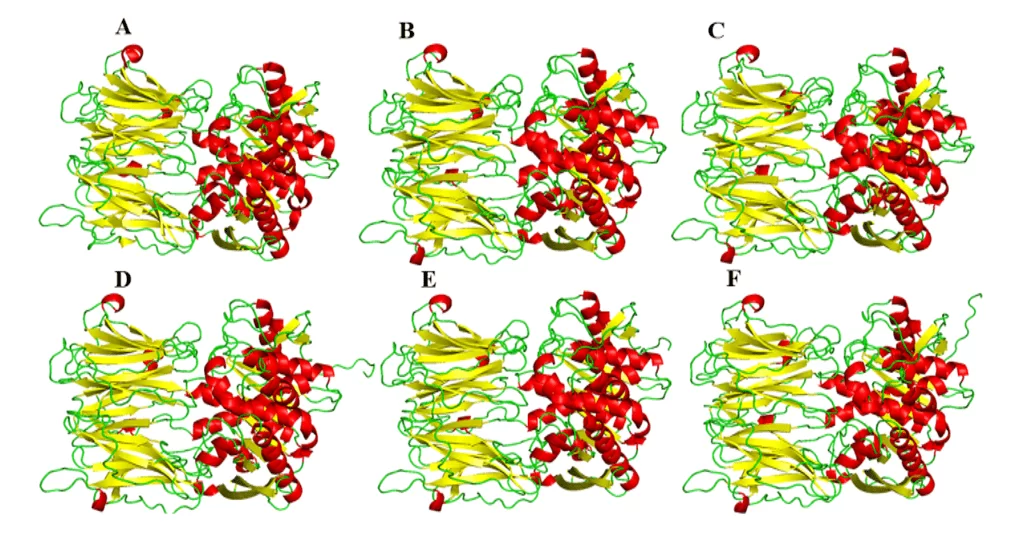
Als Ergebnis der vergleichenden Modellierung wurden die Modelle der vorliegenden Studie gewonnen, die eine große visuelle Ähnlichkeit in ihrer dreidimensionalen Form in Bezug auf die Form nahelegten (Abbildung 5). Hinsichtlich des Vergleichs zwischen den Modellen stellten sie untereinander unterschiedliche Mengen in den Strukturen von A-Helix und Blatt-A dar. Dieser Unterschied lässt sich durch die spezifischen Unterschiede in der Zusammensetzung von Proteinrückständen erklären, aber solche Unterschiede zeigten sich in wichtigen Regionen dieser Enzyme, wie der Bindungsstelle, nicht. Daher sind diese Unterschiede nicht so wichtig, um die Strukturen oder das Profil der Interaktion mit einem möglichen Medikament an der Bindungsstelle zu ändern.
Abbildung 5: Modelle und Schimmel der OPBs der Leishmanie spp., in rot die –Helixen nachgewiesen, grün die Griffe und gelb die Blätter- Para OPB, (A) L.major (molde) e modelos: (B) L. brasiliensis, (C) L. donovani, (D) L. infantum, (E) L. mexicana e (F) L. panamensis.
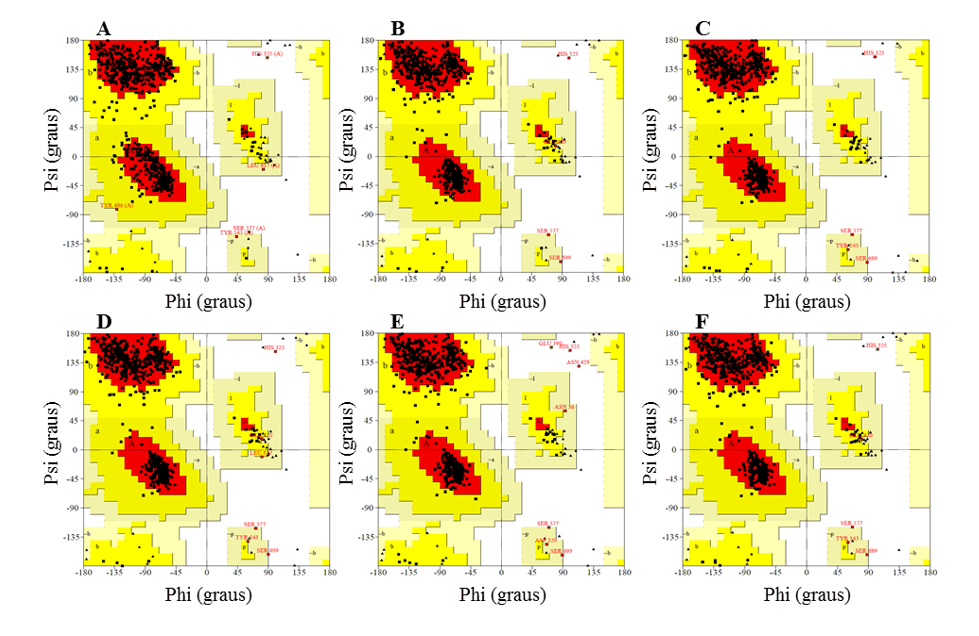
In der Validierungsphase wurden sie auf der Grundlage der Ramachandran-Graphenanalyse (bereitgestellt vom PDBsum-Server), der 3D-1D-Score (bereitgestellt durch das verify-3D-Programm) und der Score-Z (bereitgestellt von ProSA-Webserver) analysiert. Die für die Modelle ermittelten Werte wurden mit denen für die Form verglichen.
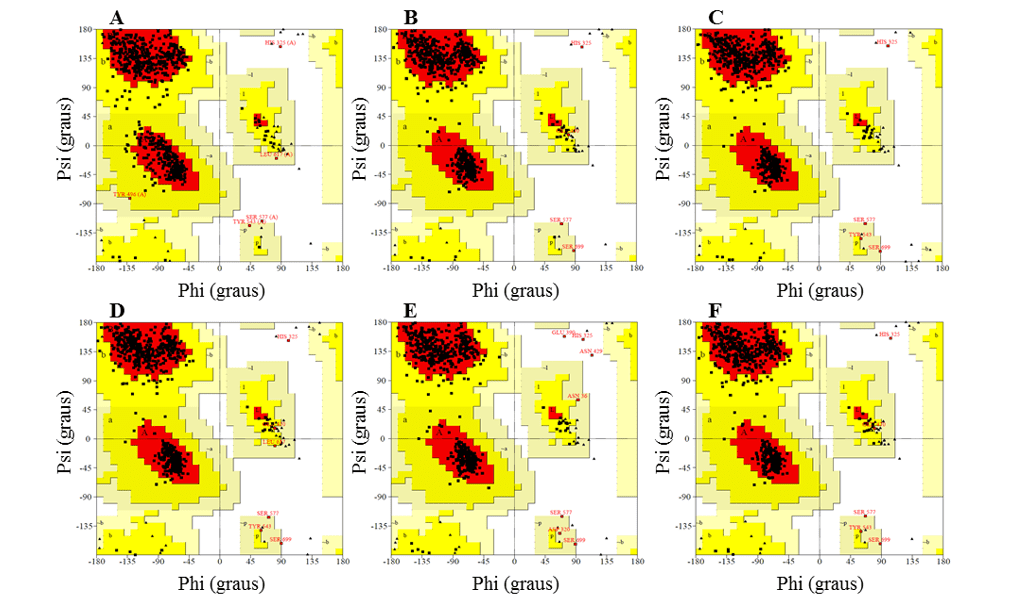
Im Ramachandran-Diagramm stellten die Modelle die meisten Rückstände in den günstigen Regionen dar, die zwischen 91,2 und 92,3 % lagen, während der Rückstandsanteil in ungünstigen Regionen maximal 0,5 % betrug und die besten Modelle L.infantum und L.brasilienses waren. Diese Modelle wiesen mit 92,3 % bzw. 92,2 % die höchste Rückstandsquote in günstigen Regionen und in ungünstigen Regionen mit 0,3 % bzw. 0,2 % die geringste Rückstandsquote auf (Abbildung 6).
In allen Modellen konnte man beobachten, dass der Ser-Rückstand, der Teil der katalytischen Triade ist, in den ungünstigen Regionen lag. Diese Tatsache hat jedoch keinen Einfluss auf die Gültigkeit der Modelle, da beim Vergleich der Form das gleiche Ergebnis vorliegt. Daher konfiguriert dieses Ergebnis keine geringe Zuverlässigkeit der Modelle.
Abbildung 6: Ergebnisse von Ramachandran-Diagrammen, die vom PROCHECK-Programm, den Strukturen der erzeugten OPB-Modelle und der Form ermittelt werden.
| Strukturen | % Abfall in Regionen | ||||
| Günstigen | Erlaubt | Ungünstigen | |||
| OLIGOPEPTIDASE B | L. major (A)
(PDB 2XE4) |
90,2 | 9,5 | 0,3 | |
| L.brasilienses (B) | 92,2 | 7,7 | 0,2 | ||
| L. donovani (C) | 91,9 | 8,0 | 0,2 | ||
| L. infantum (D) | 92,3 | 7,3 | 0,3 | ||
| L. Mexicana (E) | 91,2 | 8,3 | 0,5 | ||
| L. panamensis (F) | 91,4 | 8,3 | 0,3 | ||
Quelle: Erstellt vom Autor auf der Grundlage von Procheck-Ergebnissen.
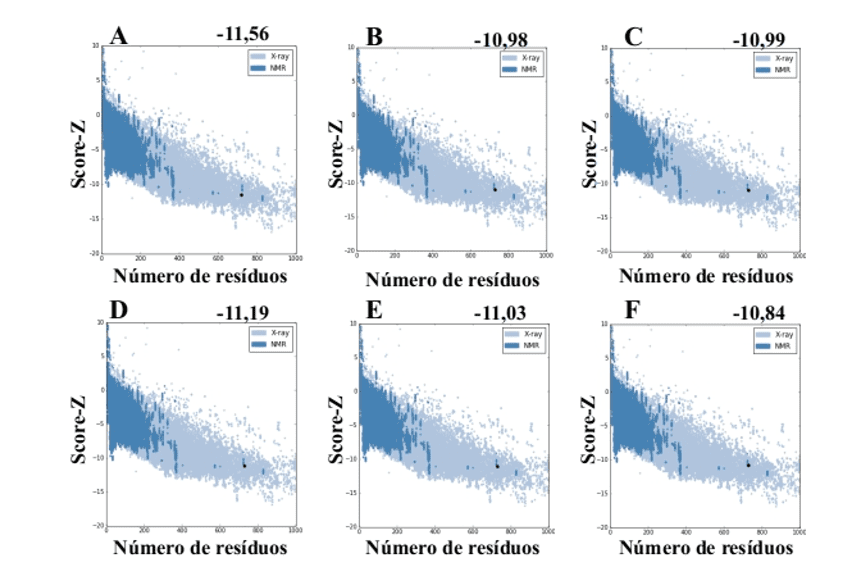
Bei Verwendung des ProSA-Webservers aus den von MODELLER generierten Modellen wurden Werte von score-Z -10.84 bis -11.19 dargestellt, und diese Werte sind mit PDB-Strukturen kompatibel (Abbildung 7).
Abbildung 7: Z-Score-Ergebnisse auf dem ProSA-Webserver der Formstrukturen berechnet (zum Vergleich). (A) L .major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. Die Region in Dunkelblau gibt die Punktzahl der Proteine an, die durch NMR und in hellblau der durch Röntgenbeugung erhaltenen Proteine erhalten wurden.
Das Verify 3D-Programm wurde verwendet, um die Kompatibilität zwischen 1D- und 3D-Strukturen zu bewerten. Die erhaltenen Modelle zeigten 95,28% bis 98,34% der Aminosäuren mit 3D-1D>-Kompatibilität 0,2, und die Modelle L. donovani und L.brasilienses erzielten bessere Ergebnisse. Gemäß den idealen Parametern des Programms sollten die meisten Rückstände Werte über Null aufweisen, da Werte unter Null Bereiche des Moleküls mit Problemen anzeigen. Der Anteil der Aminosäuren mit 3D-1D>-Kompatibilität 0,2 muss über 80% liegen (EISENBERG et al., 1997). Diese Ergebnisse deuten darauf hin, dass die Modelle eine 1D-3D-Kompatibilität aufwiesen und die Rückstände, die inkompatibel waren, nicht Teil der aktiven Stelle der Enzyme sind (Tabelle 2).
Tabelle 2: Ergebnisse von Verify 3D, die den Prozentsatz der Residuen mit der Punktzahl > 0,2 anzeigen.
| Estruturas | % de resíduos com score > 0,2 | |
| OLIGOPEPTIDASE B | L.major
(PDB 2XE4) |
93,20 |
| L.brasilienses | 95,62 | |
| L.donovani | 97,12 | |
| L.infantum | 94,93 | |
| L.mexicana | 95,62 | |
| L.panamensis | 94,93 |
Quelle: Authoral.
VALIDIERUNG DER CATALYTIC TRIAD DES ENZYMES DER MODELS
Aufgrund des Mechanismus von PB ist es wichtig, den Abstand und die Ausrichtung zwischen Aminosäurerückständen und der katalytischen Triade (Ser, Asp und His) der erzeugten Modelle zu analysieren, um die Zuverlässigkeit der Modelle zu erhöhen.
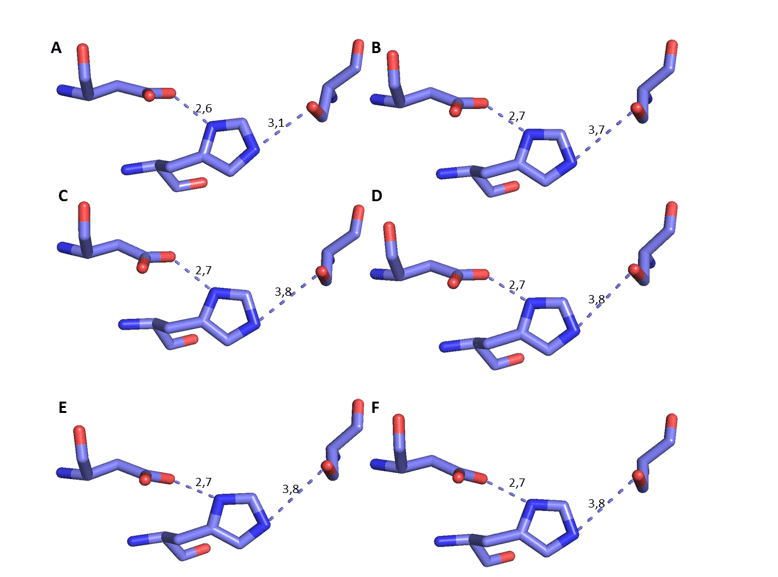
Es ist möglich, den Vergleich dieser spezifischen Rückstände der OPB-Modelle mit der betreffenden Form in Abbildung 17 zu beobachten. Darüber hinaus war es möglich, den Abstand zwischen den Rückständen der katalytischen Triade zu messen, basierend auf dem Mechanismus der Interaktion zwischen der Stelle und dem Substrat, mit Seiner als Referenz (DEREWENDA et al. , 1994). Daher muss es einen besonderen Abstand zum Wirkmechanismus geben. Dieser Abstand sollte etwa 3,5 % zwischen seinem Sauerstoff und SerStickstoff betragen, zusätzlich zu dem Abstand von etwa 2,6 % zwischen Aspartatsauerstoff und seinem anderen Stickstoff, wie in der Literatur beschrieben (DEREWENDA et al. , 1994). In dieser Analyse konnte überprüft werden, ob der betreffende Abstand geringfügig abgenommen hat und immer noch dem entspricht, was bereits beschrieben wurde. Dieser Abstand ist wichtig, da Serino-Proteasen seine Substratspaltung erfordern (HEDSTROM, 2002) (Abbildung 8). Basierend auf diesen zuverlässigen Ergebnissen wurde die weitere Charakterisierung der Modelle fortgesetzt.
Abbildung 8: 3D-Darstellung der katalytischen Triade der OPBs bzw. Ser, His und Asp. Die dunkelblaue Farbe stellt das Stickstoffatom, den roten Sauerstoff und den fliederfarbenen Kohlenstoff dar. Para OPB, (A) L.major (molde)e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis.
CHARAKTERISIERUNG VON OLIGOPEPTIDASES VON LEISHMANIAS
PREDICTION SEKUNDÄRE STRUKTUR
Aus der Vorhersage der sekundären Struktur von OPBs-Enzymen wurden Anzahl und Position der Sekundärstrukturen der Modelle aufgedeckt. Die PSISPRED präsentierte zwischen 15 und 16-Helix-Strukturen und alle präsentierten 38 Paare von A-Blättern (Tabelle 3). Aus den von PSIPRED generierten Ergebnissen wurde mit dem Pymol-Programm ein visueller Vergleich dieser vorhergesagten Sekundärstruktur mit den dreidimensionalen Strukturen der 5 Modelle durchgeführt. Die Modelle von L.brasilienses und L. panamensis zeigten die beste Ähnlichkeit, in Bezug auf die sekundären und tertiären vorhergesagten Strukturen (2D-3D), die 15 -Helixen und 38 -Blätter aufweisen. Von den 15-S-Propellern, die in der dreidimensionalen Struktur des Modells 8 der Propeller dargestellt wurden, befanden sich sie in der gleichen Position wie vom PSIPRED vorhergesagt. Diese Helixen entsprechen der Aminosäuresequenz: 59 bis 74, 78 bis 93, 535 bis 540, 545 bis 563, 631 bis 640, 703 bis 721 und 727 bis 730.
In der allgemeinen Analyse gab es keinen verschärften Unterschied in den Mengen von A-Helix und Blättern zwischen den Vorhersagen und den erhaltenen Modellen (Tabelle 3).
Tabelle 3: Vergleich zwischen den von Psipred vorhergesagten sekundären Strukturen und denen, die durch Pymol gefunden wurden.
| Modelos | α-Hélices | Folhas β | PSIPRED
α-Hélices |
PSIPRED
Folhas β |
| L.brasiliensis | 15 | 38 | 11 | 36 |
| L. donovani | 16 | 38 | 11 | 36 |
| L. infantum | 16 | 38 | 11 | 36 |
| L. mexicana | 15 | 38 | 10 | 36 |
| L. panamensis | 15 | 38 | 11 | 36 |
Quelle: Authoral.
Außerdem konnte festgestellt werden, dass die Menge von 15 Propellern für L.brasilienses, L. Mexicana und L. panamensis gleich war. Genau wie L.infantum und L.donovani bekamen sie die Zahl 16 -Helix. Nach dem phylogenetischen Baum befinden sich diese beiden genannten Gruppen im selben internen Knoten und gelten als monophyletisch (Abbildung 2).
Anschließend wurde die RMSD (Root-Mean-Quadrat-Abweichung) zwischen den Modellen und der Form durchgeführt. Die Werte waren vielversprechend, wie aus Tabelle 4 hervorgeht, da die RMSDs den Wert von 0,19 ° nicht überstiegen. Diese Feststellung kann durch den hohen Grad an Identität zwischen der Form und den jeweiligen Modellen gerechtfertigt werden. Im Allgemeinen wird erwartet, daß Proteine mit einer Identität von mehr als 30 % eine ausgezeichnete Überlappung der Hauptketten aufweisen und so eine RMSD in der Größenordnung von 2 % erhalten (BENNER et al., 1997 ; CHOTHIA et al., 1986).
Tabelle 4: RMSDs der vom Modeller erzeugten OPBs, mit Orientierung die Alphakohlenstoffe der OPB-Form von L. Major.
| Molde | Modelos | RMSD (Å) |
| L. major
(2XE4) |
L.brasiliensis | 0,15 |
| L. donovani | 0,15 | |
| L. infantum | 0,16 | |
| L. Mexicana | 0,19 | |
| L. panamensis | 0,14 |
Quelle: Authoral.
MAP VON MOLECULAR ELECTROSTATIC POTENTIAL (MEP) DES SURFACE OF ENZYMES AND RECEPTIVE SITES
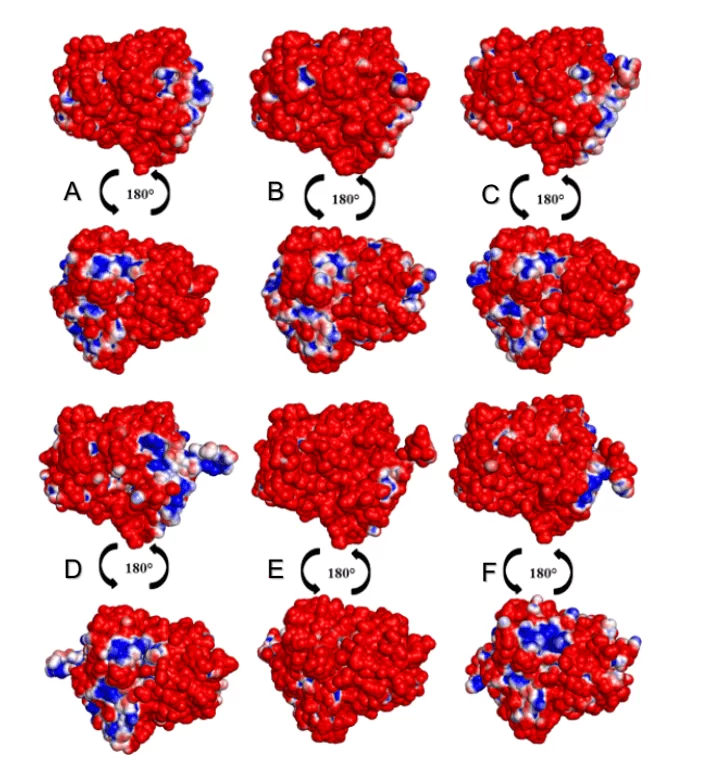
Bei der Analyse der MDPs der Oberflächen der OPBs konnte festgestellt werden, dass alle Leishmanie-Arten einen höheren Prozentsatz negativer als positiver Regionen aufwies, wie in Abbildung 15 dargestellt. Die Arten L. donovani und L. infantum (grüne Gruppe) schlugen einen negativen Bereich (in blauer Farbe) in derselben Region vor. Die Arten L.brasilienses und L. panamensis (rote Gruppe) präsentierten eine ähnliche negative Region in einem ähnlichen farbmetrischen Muster. Beide Ergebnisse können dadurch gerechtfertigt werden, dass die Arten im Vergleich zueinander zum gleichen Monophyletikgehören gehören (Abbildung 2).
Abbildung 9: Karte des elektrostatischen Potentials der 3D-Modelle von Leishmania spp OPBs und deren Form. Para OPB, (A) L.major (molde) e modelos: (B) L. brasilienses (C) L. donovani, (D) L. infantum, (E) L. mexicana (F) L. panamensis. In der blauen Farbe präsentiert es den positiven Bereich und in der roten Farbe den negativen Bereich.
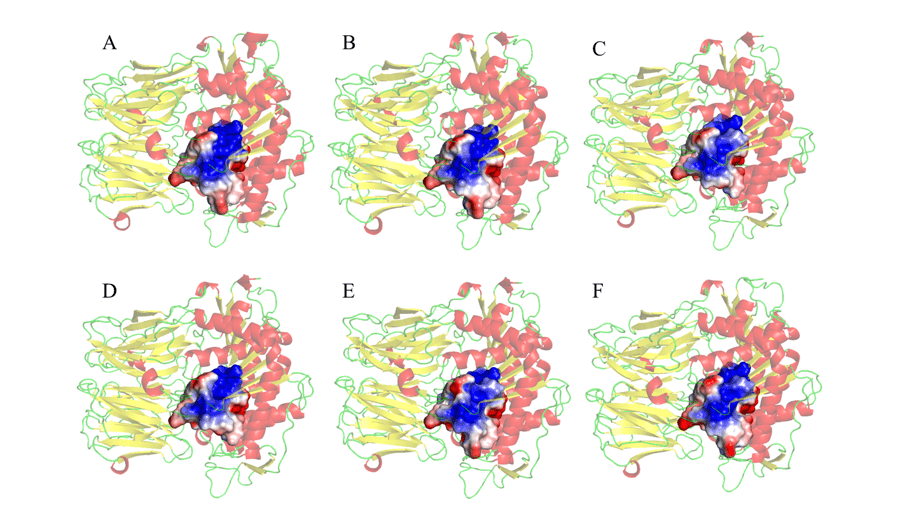
Schließlich wurde der MEP der Rückstände auch in einem Radius von 5 ° um die katalytische Triade durchgeführt, wie in Abbildung 10 dargestellt. Die Wahl, das MEP der Region um die katalytische Triade zu beobachten, lag daran, dass dies die zweite Region der Interaktion zwischen dem Enzym und dem Substrat ist, in der es Eine Unterkunft geben wird (ANDERSSON et al., 2010). So war es im Hinblick auf eines der Ziele der Studie möglich, signifikante Ähnlichkeiten und Übereinstimmungen zwischen der Form und den getesteten Modellen zu überprüfen. Diese Ergebnisse, wie in Abbildung 10 dargestellt, zeigten hauptsächlich einen größeren elektropositiven Anteil (in blau) im zentralen Bereich der Anschlussstellen. Negative Regionen (rot) wurden in Randgebieten der untersuchten MdEP beobachtet. Diese Ergebnisse sind vielversprechend, da festgestellt wurde, dass diese Regionen in allen Modellen völlig ähnlich sind, und kann bei der Entwicklung eines Medikaments helfen, das in allen untersuchten Modellen spezifisch wirken kann.
Abbildung 10: Darstellung der potenziellen Karte der elektrostatischen Oberfläche von Aminosäurerückständen, 5 um die katalytische Triade, die die aktive Stelle des Enzyms bilden. (A) L.major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. In rot sind die ‘-Helixen, grün die Griffe und gelb die Blätter-‘. In der goldenen Farbe die Bindungsseite. In der blauen Farbe präsentiert es den positiven Bereich und in der roten Farbe den negativen Bereich.
DOGSITESCORER
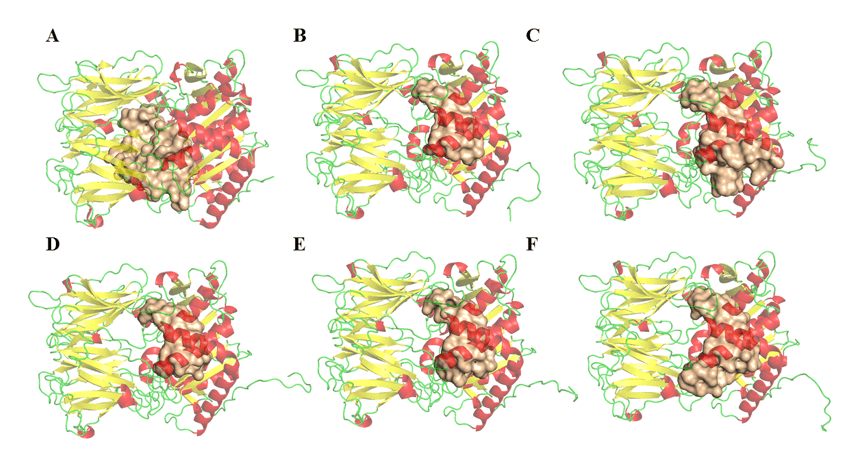
Die Bestimmung der Volumen-, Flächen- und Tiefenparameter der möglichen Bindungsstellen von Opb-Enzymen der Leishmania-Arten wurde im DoGSiteScorer (http://poseview.zbh.uni-hamburg.de/)-Programm von ProteinsPlus – Structure-Based Modeling Support Server (VOLKAMER et al., 2012) durchgeführt. Das Programm weist auf drei Link-Sites hin, wie in der Literatur beschrieben. Abbildung 11 zeigt den Vergleich zwischen dem möglichen Bereich des aktiven Standorts und seinen Unterschieden. Diese Variablen unter den OPBs können mit Rückständen zusammenhängen, die nicht als wichtig für ihre Hemmung beschrieben werden, aber während dieser Analyse berücksichtigt wurden. Somit schließt dieses Ergebnis nicht aus, dass Antipain oder ein anderes Molekül ein breites Spektrum an Hemmung auf das Enzym hat, da die Ergebnisse im Zusammenhang mit drugscore in allen Regionen sehr ähnlich und positiv waren. Tabelle 5 wird beobachtet, eine relative Diskrepanz zwischen volumen- und flächenmäßigen Ergebnissen der in den Modellen gefundenen Hohlräume. Dieses Ergebnis deutet nicht auf eine Voreingenommenheit des Zweifels über das hemmende Potenzial eines einzelnen Medikaments in den jeweiligen Enzymen hin. Es achtet jedoch darauf, dass einige Proteine größere Hohlräume haben als andere. Allerdings haben dieselben Hohlräume gemeinsam, und dies sind die Rückstände, die die katalytische Stelle bilden, wie in Abbildung 11 zu sehen ist und untersucht werden kann.
Tabelle 5: Werte, die sich auf die möglichen Verbindungsbereiche der OPBs beziehen (vom Server erhalten).
| Estruturas | DogSiteScoore | |||
| Volume | Área | Drug Score | ||
| OLIGOPEPTIDASE B | L. major
(PDB 2XE4) |
1690,62 | 1818,41 | 0,80 |
| L.brasiliensis | 1527,84 | 1766,63 | 0,80 | |
| L. donovani | 1074,57 | 1428,55 | 0,79 | |
| L. infantum | 1309,97 | 1572,19 | 0,80 | |
| L. mexicana | 800,92 | 799,38 | 0,85 | |
| L. panamensis | 971,96 | 1083,40 | 0,81 | |
Quelle: Authoral.
Abbildung 11: OPBs-Strukturen und mögliche Verbindungsbereiche (vom DogSite-Server erhalten). (A) L .major (molde)e modelos: (B) L. brasilienses, (C), L. donovani, (D) L. infantum, (E) L. mexicana e (F) L. panamensis. In rot sind die ‘-Helixen, grün die Griffe und gelb die Blätter-‘. In der goldenen Farbe die Bindungsseite.
NORMAL MODES
Nach der Charakterisierung des Enzyms in den strukturellen, oberflächlichen und bindenden Stellen wurden die normalen Modi für die Enzyme jeder Spezies mit dem Zweck ihrer jeweiligen Bewegungen durchgeführt.
Nach Entspannungs- und Energie-Minimiza-Es, die durch molekulare Dynamik in GROMACS durchgeführt wurden, wurden diese Strukturen normalen Analysen unterzogen, um mögliche kompatible Bewegungen zu beobachten.
Es war möglich, in allen untersuchten Modellen eine expressive Bewegung einer bestimmten Helix zu beobachten, und bei der Analyse dieser Region in ihrer Aminosäurezusammensetzung ist es möglich, zu bemerken, dass sie hochkonserviert ist. So wurde das Bewegungsmuster in allen fraglichen Modellen wiederholt. Diese Region schlug eine lineare Bewegung vor, die sich vom Zentrum weg zur Peripherie bewegte und die katalytische Triade freilegte. Dies kann auf die Bewegung des Proteins für die Anpassung des Substrats hinweisen.
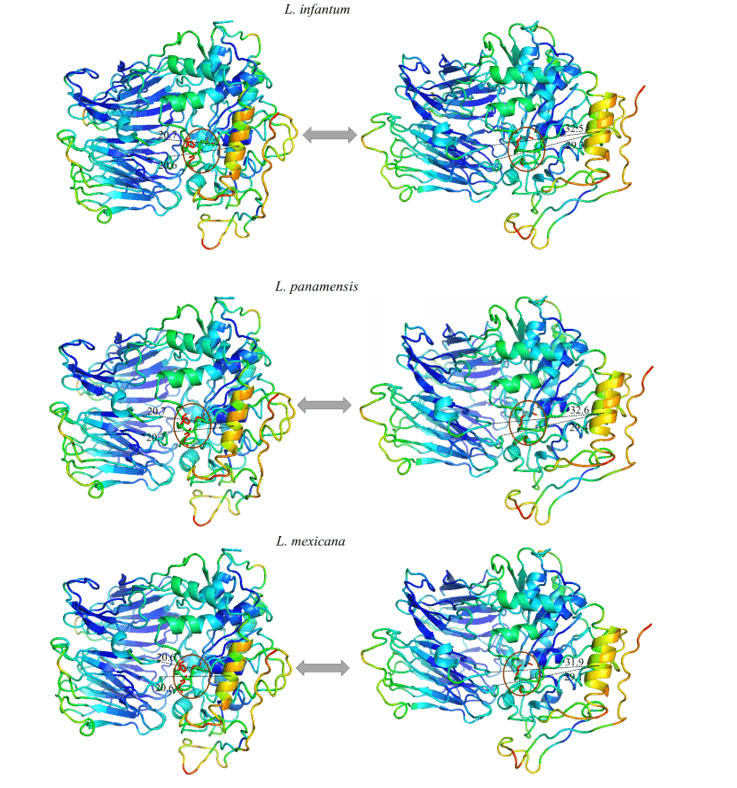
Bei der Beobachtung der Abbildungen 12 und 13 kann in allen Modellen der Studie das gleiche Farbmuster beobachtet werden. Diese Daten stellen die Fähigkeit dar, dass sich die Region in eine bestimmte Richtung bewegen muss. Die blauen Farben stellen während der Simulation durch normale Modi unvariablere Bereiche dar, wobei ein Steifigkeitsmuster in den Proteinen vor allem den Blättern der ‘-Propellerdomäne und einigen ‘-Helixen, die die katalytische Domäne bilden, entspricht. Farben in der Nähe von Grün stellen Zwischenregionen in Bezug auf Kapazität und Bewegungsumfang dar. Auf der Grundlage dieser Informationen ist es möglich, diese Färbung in Schleifen an den Enden der Modelle sowie in einigen ‘-Helixen der katalytischen Domäne wahrzunehmen. Schließlich gibt es jene Regionen, die eine orange/rote Färbung präsentierten, in denen die Darstellung eines riesigen Bewegungspotenzials ist. So kann man sehen, dass es kleine Bereiche von Schleifen an den Enden der katalytischen Domänen mit diesem Bewegungspotential gibt, sowie eine ‘-Helix, die sich im Teil der katalytischen Domäne befindet, genauer gesagt vor der katalytischen Triade der OPBs, in der sie die ausdrucksvollste Bewegung der Studie erhielt.
Alle Modelle erhielten die gleiche Bewegungskonfiguration, wobei sich eine Helix (in Orange) als die Region mit der höchsten Bewegungskapazität erwies. So erhielten die OPBs von L. brasiliensis, L. donovani, L. infantum, L. mexicana und L. panamensis in ihrem größten Bewegungsbereich jeweils 9,8 , 11,3 , 11,8, 11,9 und 11,6 ” (Abbildung 12 und 13).
Abbildung 12: Ergebnis der Analyse durch normale Darstellungsmodi durch das Pymol-Programm, im Bild sind die Bewegungen der Modelle in ihren entspannten Formen (rechts) im Vergleich zur Bewegung größerer Amplitude (links) dargestellt. Ebenfalls vertreten sind die starrsten Regionen (Dunkelblau), Regionen mit wenig Bewegung (Hellblau), Zwischenregionen (Grün), Regionen mit gutem Bewegungspotenzial (Orange) und extrem formbare Regionen (rot) (Teil 1/2).
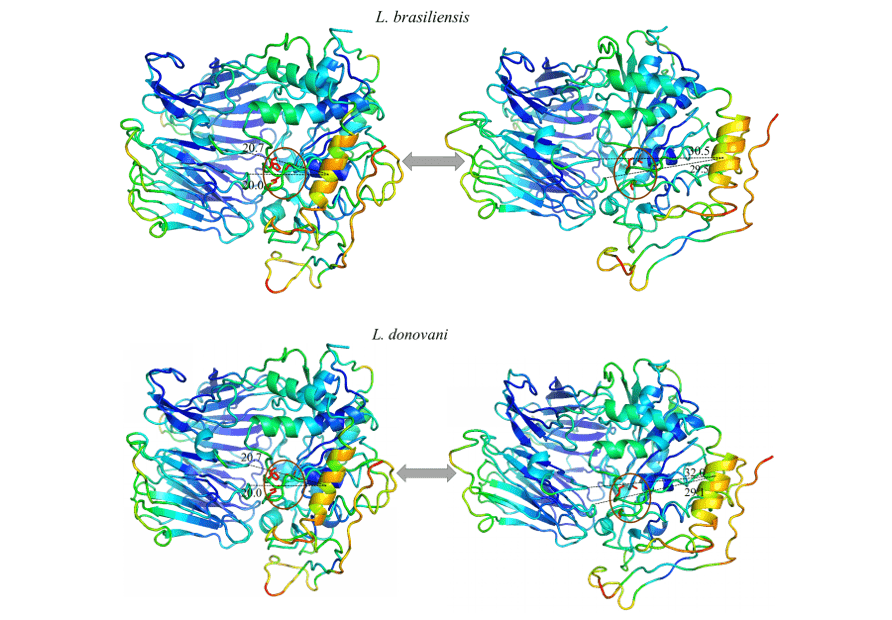
Abbildung 13: Ergebnis der Analyse durch normale Darstellungsmodi durch das Pymol-Programm, im Bild sind die Bewegungen der Modelle in ihren entspannten Formen (rechts) im Vergleich zur Bewegung größerer Amplitude (links) dargestellt. Ebenfalls vertreten sind die starrsten Regionen (Dunkelblau), Regionen mit wenig Bewegung (Hellblau), Zwischenregionen (Grün), Regionen mit gutem Bewegungspotenzial (Orange) und extrem formbare Regionen (rot). (Teil 2/2)
Vor diesem Grund befürworten normale Studien einen möglichen Mechanismus von OPBs, die für Leishmanie-Spp-Arten noch nicht beschrieben sind. bis zur vorliegenden Studie.
FAZIT
In dieser Studie wurden die dreidimensionalen Modelle des Opb-Enzyms von L.brasiliensis, L. donovani, L. infantum, L. mexicana und L. panamensis erhalten. Die Validierung der Modelle präsentierte zuverlässige Ergebnisse für alle erhaltenen dreidimensionalen Modelle. Bei der Charakterisierung des Enzyms zeigte die oberflächenelektrostatische Potentialkarte, dass die meisten Rückstände eine negative Ladung aufwiesen. Bei der Charakterisierung der Region um die katalytische Triade zeigte sie Ähnlichkeit zwischen Volumen, Fläche und Entsprechung zwischen positiven und negativen Rückständen. Daher konnte überprüft werden, ob die Ergebnisse der Analyse durch normale Modi eine expressive Bewegung in einer der spezifischen Helixen nahelegten, die einen linearen Abstand davon vom Zentrum zur Peripherie einlegten und so die katalytische Triade freilegten. Die Beschreibung dieser Bewegungen, die von diesem Enzym durchgeführt werden, ist von großer Bedeutung, um das Verständnis seiner Funktionsweise zu unterstützen.
Schließlich können die Ergebnisse dieser Studie der wissenschaftlichen Gemeinschaft Wissen hinzufügen, indem sie Erläuterungen und neue Fragen im Zusammenhang mit dem Thema bringen und als Grundlage für eventuelle Studien im Gesundheitsbereich dienen.
REFERENZEN
A. Benner S, Cannarozzi G, Gerloff D, Turcotte M, Chelvanayagam G. Bona Fide Predictions of Protein Secondary Structure Using Transparent Analyses of Multiple Sequence Alignments. Chem Rev. 1997;97(8):2725-2844. doi:10.1021/cr940469a.
Alva, V., Nam, S. Z., Söding, J., & Lupas, A. N. (2016). The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Research, 44(W1), W410–W415. doi.org/10.1093/nar/gkw348.
Alvarenga DG, Escalda PMF, da Costa ASV, Monreal MTFD. Leishmaniose visceral: Estudo retrospectivo de fatores associados à letalidade. Rev Soc Bras Med Trop. 2010;43(2):194-197.
Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389‐3402. doi:10.1093/nar/25.17.3389.
Andersson CD, Chen BY, Linusson A. Mapping of ligand-binding cavities in proteins [published correction appears in Proteins. 2011 Apr;79(4):1363]. Proteins. 2010;78(6):1408–1422. doi:10.1002/prot.22655.
Bailey F, Mondragon-Shem K, Hotez P, et al. A new perspective on cutaneous leishmaniasis-Implications for global prevalence and burden of disease estimates. PLoS Negl Trop Dis. 2017;11(8):e0005739. Published 2017 Aug 10. doi:10.1371/journal.pntd.0005739.
Carmo RF, Luz ZMP da, Bevilacqua PD. Percepções da população e de profissionais de saúde sobre a leishmaniose visceral. Cien Saude Colet. 2016;21(2):621-628. doi:10.1590/1413-81232015212.10422015.
Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5(4):823‐826.
Derewenda ZS, Derewenda U, Kobos PM. (His)Cε-H···O=C< Hydrogen Bond in the Active Sites of Serine Hydrolases. J Mol Biol. 1994;241(1):83-93. doi:10.1006/JMBI.1994.1475.
Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32(Web Server issue):W665–W667. doi:10.1093/nar/gkh381.
Eisenberg, D., Lüthy, R., & Bowie, J. U. (1997). [20]VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods in Enzymology, 277, 396–404. https://doi.org/10.1016/S0076-6879(97)77022-8.
Eyal E, Lum G, Bahar I. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics. 2015;31(9):1487–1489. doi:10.1093/bioinformatics/btu847.
Ghorbani Masoud, Farhoudi Ramin. Leishmaniasis in humans: drug or vaccine therapy? Drug Des Devel Ther. 2018;12:25-40. doi:10.2147/DDDT.S146521.
Hedstrom L. Serine Protease Mechanism and Specificity. Chem Rev. 2002;102(12):4501-4524. doi:10.1021/cr000033x.
Katsila T, Spyroulias GA, Patrinos GP, Matsoukas MT. Computational approaches in target identification and drug discovery. Comput Struct Biotechnol J. 2016;14:177–184. Published 2016 May 7. doi:10.1016/j.csbj.2016.04.004.
Kumar S, Tamura K, Nei M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5(2):150-163.
Macedo-Silva RM, dos Santos C de LP, Diniz VA, De Carvalho JJ, Guerra C, Côrte-Real S. Peripheral blood fibrocytes: New information to explain the dynamics of Leishmania infection. Mem Inst Oswaldo Cruz. 2014;109(1):61-69. doi:10.1590/0074-0276130247
Machado P de A, Carneiro MPD, Sousa-Batista A de J, et al. Leishmanicidal therapy targeted to parasite proteases. Life Sci. 2019;219:163-181. doi:10.1016/J.LFS.2019.01.015.
Morris, A. L., MacArthur, M. W., Hutchinson, E. G., & Thornton, J. M. (1992). Stereochemical quality of protein structure coordinates. Proteins: Structure, Function, and Bioinformatics, 12(4), 345–364. https://doi.org/10.1002/prot.340120407.
Ovchinnikova M V., Mikhailova AG, Karlinsky DM, Gorlenko VA, Rumsh LD. Reversible cyclic thermal inactivation of oligopeptidase B from Serratia proteamaculans. Acta Naturae. 2018;10(2):65-70.
Ramachandran, G. N., Ramakrishnan, C., & Sasisekharan, V. (1963). Stereochemistry of polypeptide chain configurations. Journal of Molecular Biology, 7(1), 95–99. https://doi.org/10.1016/S0022-2836(63)80023-6.
Santos Filho, O. A., & Alencastro, R. B. de. (2003). Modelagem de proteínas por homologia. Química Nova, 26(2), 253–259. https://doi.org/10.1590/S0100-40422003000200019
Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. Published 2011 Oct 11. doi:10.1038/msb.2011.75.
SODERO ACR, DOS SANTOS ACGO, MELLO JFRE, et al. Oligopeptidase B and B2: comparative modelling and virtual screening as searching tools for new antileishmanial compounds. Parasitology. 2017;144(4):536-545. doi:10.1017/s0031182016002237.
Swenerton RK, Zhang S, Sajid M, et al. The oligopeptidase B of Leishmania regulates parasite enolase and immune evasion. J Biol Chem. 2011;286(1):429-440. doi:10.1074/jbc.M110.138313.
Volkamer, A., Kuhn, D., Rippmann, F., & Rarey, M. (2012). Dogsitescorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics, 28(15), 2074–2075. https://doi.org/10.1093/bioinformatics/bts310.
Wang Q, Arighi CN, King BL, et al. Community annotation and bioinformatics workforce development in concert–Little Skate Genome Annotation Workshops and Jamborees. Database (Oxford). 2012;2012:bar064. Published 2012 Mar 20. doi:10.1093/database/bar064
Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics. 2016;54:5.6.1–5.6.37. Published 2016 Jun 20. doi:10.1002/cpbi.3.
Wiederstein, M., & Sippl, M. J. (2007). ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research, 35(SUPPL.2), 407–410. https://doi.org/10.1093/nar/gkm290.
WHO. Integrating Neglected Tropical Diseases into Global Health and Development: Fourth WHO Report on Neglected Tropical Diseases.; 2017. http://apps.who.int/iris/bitstream/10665/255011/1/9789241565448-eng.pdf?ua=1.
WHO. (2019). Leishmanioses – Informe Epidemiológico das Américas No 7 – Março, 2019. Retrieved from http://iris.paho.org/xmlui/bitstream/handle/123456789/50505/ 2019-cde-leish-informe-epi-das-americas.pdf?sequence=2&isAllowed=y.
ANHANG – ZAHLEN UND TABELLEN IN ENGLISCHER SPRACHE
Schema 1: Vereinfachtes Schema der Materialschritte und -methoden.
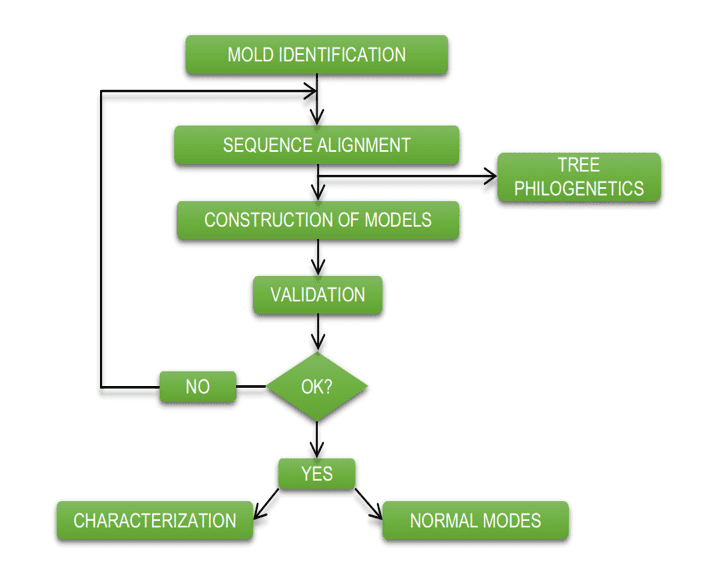
Tabelle 1: Prozentsatz der Identität zwischen den Modellen der Oligopeptität B der Leishmanischen Art und ihrer jeweiligen Form.
| Mold protein (code PDB) | Models OPB
(código uniprot) |
Identity (%) | Gaps (%) |
| OPB
L. major (code PDB 2XE4) |
L.brasiliensis
(A4H5Q8) |
86 | 0 |
| L. donovani
(C9EF60) |
96 | 0 | |
| L. infantum
(A4HTZ8) |
96 | 0 | |
| L. Mexicana
(E9AMS8) |
90 | 0 | |
| L. panamensis
(A0A088RJA7) |
86 | 0 |
Fonte: Autoral.
Abbildung 6: Ergebnisse von Ramachandran-Diagrammen, die vom PROCHECK-Programm, den Strukturen der erzeugten OPB-Modelle und der Form ermittelt werden.
| Structures | % waste in the regions | ||||
| Favorable | Allowed | Unfavorable | |||
| OLIGOPEPTIDASE B | L. major (A)
(PDB 2XE4) |
90,2 | 9,5 | 0,3 | |
| L.brasilienses (B) | 92,2 | 7,7 | 0,2 | ||
| L. donovani (C) | 91,9 | 8,0 | 0,2 | ||
| L. infantum (D) | 92,3 | 7,3 | 0,3 | ||
| L. Mexicana (E) | 91,2 | 8,3 | 0,5 | ||
| L. panamensis (F) | 91,4 | 8,3 | 0,3 | ||
Quelle: Erstellt vom Autor auf der Grundlage von Procheck-Ergebnissen.
Abbildung 7: Z-Score-Ergebnisse auf dem ProSA-Webserver der Formstrukturen berechnet (zum Vergleich). (A) L .major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. Die Region in Dunkelblau zeigt die Punktzahl der Proteine an, die durch NMR und in hellblau der durch Röntgenbeugung erhaltenen Proteine erhalten wurden.
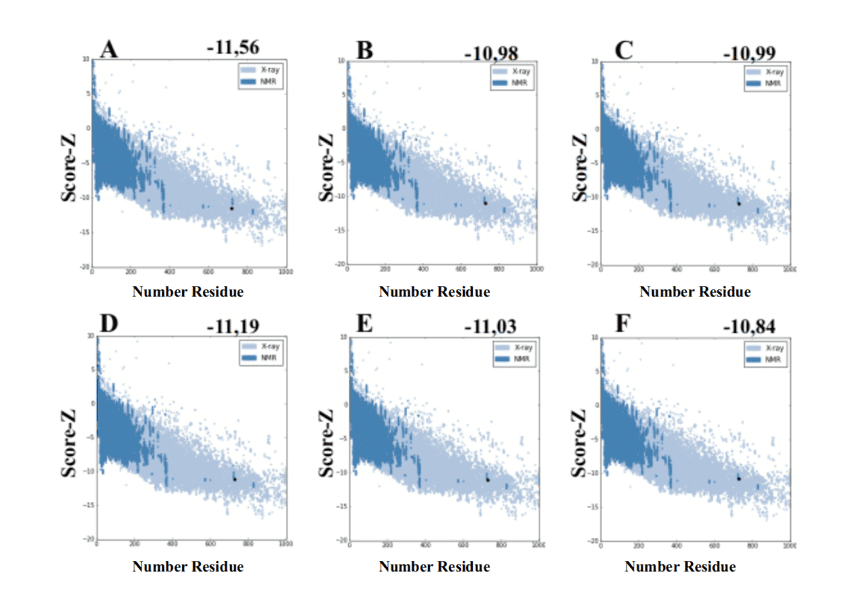
Tabelle 2: Ergebnisse von Verify 3D, die den Prozentsatz der Residuen mit der Punktzahl > 0,2 anzeigen.
| Structures | % residue with score > 0,2 | |
| OLIGOPEPTIDASE B | L.major
(PDB 2XE4) |
93,20 |
| L.brasilienses | 95,62 | |
| L.donovani | 97,12 | |
| L.infantum | 94,93 | |
| L.mexicana | 95,62 | |
| L.panamensis | 94,93 |
Quelle: Authoral.
Tabelle 3: Vergleich zwischen den von Psipred vorhergesagten sekundären Strukturen und denen, die durch Pymol gefunden wurden.
| Models | α-Hélix | Sheet β | PSIPRED
α-Hélix |
PSIPRED
Sheet β |
| L.brasiliensis | 15 | 38 | 11 | 36 |
| L. donovani | 16 | 38 | 11 | 36 |
| L. infantum | 16 | 38 | 11 | 36 |
| L. mexicana | 15 | 38 | 10 | 36 |
| L. panamensis | 15 | 38 | 11 | 36 |
Quelle: Authoral.
Tabelle 4: RMSDs der vom Modeller erzeugten OPBs, mit Orientierung die Alphakohlenstoffe der OPB-Form von L. Major.
| Mold | Models | RMSD (Å) |
| L. major
(2XE4) |
L.brasiliensis | 0,15 |
| L. donovani | 0,15 | |
| L. infantum | 0,16 | |
| L. mexicana | 0,19 | |
| L. panamensis | 0,14 |
Quelle: Authoral.
Tabelle 5: Werte, die sich auf die möglichen Verbindungsbereiche der OPBs beziehen (vom Server erhalten).
| Structures | DogSiteScoore | |||
| Volume | Area | Drug Score | ||
| OP OLIGOPEPTIDASE B | L. major
(PDB 2XE4) |
1690,62 | 1818,41 | 0,80 |
| L.brasiliensis | 1527,84 | 1766,63 | 0,80 | |
| L. donovani | 1074,57 | 1428,55 | 0,79 | |
| L. infantum | 1309,97 | 1572,19 | 0,80 | |
| L. mexicana | 800,92 | 799,38 | 0,85 | |
| L. panamensis | 971,96 | 1083,40 | 0,81 | |
Quelle: Authoral.
[1] Biomedizinischer Abschluss in Medizinischer Biochemie.
[2] Master in Pharmazie und Pharmazie von UFRJ.
[3] Doktortitel in Chemie, Master in Organischer Chemie und Industrieller Pharmazie.
Eingesandt: Mai 2020.
Genehmigt: Mai 2020.