ARTIGO ORIGINAL
RIBEIRO, Fernando de Sá [1], JESUS, Jéssica Barbosa de [2], SOUZA, Alessandra Mendonça Teles de [3]
RIBEIRO, Fernando de Sá. JESUS, Jéssica Barbosa de. SOUZA, Alessandra Mendonça Teles de. Análise e caracterização de um promissor alvo terapêutico identificado em Leishmania spp. Revista Científica Multidisciplinar Núcleo do Conhecimento. Ano 05, Ed. 05, Vol. 09, pp. 99-132. Maio de 2020. ISSN: 2448-0959, Link de acesso: https://www.nucleodoconhecimento.com.br/saude/alvo-terapeutico, DOI: 10.32749/nucleodoconhecimento.com.br/saude/alvo-terapeutico
RESUMO
A Leishmaniose é uma doença negligenciada causada por protozoários do gênero Leishmania spp., o qual atinge cerca de 1,6 milhão indivíduos a cada ano sendo que 500 mil se apresentam na forma visceral. No Brasil ocorrem cerca de 30.000 novos casos a cada ano. Além disso, o país é responsável por 90% dos casos notificados de Leishmaniose Visceral, sendo essa forma mais grave da doença. Aliado a esses fatos, o tratamento vigente se mostra ineficaz, contribuindo para o estabelecimento de cepas resistentes. Atualmente, o tratamento apresenta diversos efeitos colaterais e danos permanentes à saúde dos pacientes, tal fato têm contribuído para a procura de novos fármacos contra a leishmaniose. A enzima oligopeptidase B (OPB) tem sido estudada como possível alvo terapêutico no desenvolvimento de agentes antiparasitários. Desta forma, o objetivo desse trabalho é construir o modelo tridimensional da enzima Oligopeptidase B de diferentes espécies de Leishmania spp. e compara-las entre si. Para tal, foi utilizado o método de modelagem comparativa. Nesse método foi feita a construção dos modelos das espécies L. brasiliensis, L. donovani, L. infantum, L. mexicana e L. panamensis, utilizando o programa MODELLER. Uma vez com os modelos prontos foram realizados o processo de validação dos mesmos e subsequentemente caracterizados, o qual foi possível constatar um grau de semelhança promissor entre os modelos. Por fim, tais modelos foram submetidos ao método de análise por modos normais, os quais obtiveram um padrão de movimento semelhante, com isso foi possível constatar um movimento em uma região específica de uma alfa-hélice, levando consequentemente a tríade da enzima se mostrar exposta, podendo ser indicativo de um mecanismo de ação. Por fim, espera-se utilizar os modelos construídos para auxiliar no desenvolvimento de uma nova terapia promissora para o tratamento da leishmaniose.
Palavras-chave: Leishmaniose, oligopeptidase B, modelagem molecular, modos normais.
INTRODUÇÃO
A leishmaniose, causada pelos protozoários Leishmania spp., é uma doença caracterizada por diversos tipos de manifestações, desde mais leves, no qual há relatos de pequenas lesões que mesmo sem o devido tratamento regridem até as mais graves como é o caso da Leishmaniose Visceral (LV). No Brasil, a forma mais grave desta doença apresenta dados alarmantes, em comparação aos outros países, fazendo do país o maior detentor dos casos de LV em toda a América (ALVARENGA, 2010; WHO, 2019).
Esta doença pertence ao grupo das doenças negligenciadas, as quais fazem parte todas aquelas doenças que atingem principalmente os países subdesenvolvidos e regiões mais pobres, de modo que não desperte o interesse no desenvolvimento de medicamentos. Logo, é necessário que sobrevenham técnicas eficientes e de baixo custo para contornar essa falta de incentivos financeiros para o estudo dessa doença. Sendo assim, podem ser utilizados métodos computacionais com finalidade de diminuir do tempo no desenvolvimento de uma nova terapia promissora e consequentemente o custo em comparação aos métodos mais tradicionais para o desenvolvimento de fármacos (BAILEY et al., 2017; WHO, 2017).
Apesar do pouco investimento nessa área, há tratamento para a doença como os antimoniais pentas valentes (fármaco de primeira escolha) ou a anfotericina B (fármaco de segunda escolha). Entretanto, tais tratamentos apresentam diversas desvantagens como a alta taxa de resistência e a grande variedade de efeitos colaterais, que vão desde enjoo até possíveis problemas causados ao terceiro (3°) par do nervo craniano, levando a dificuldades motoras (MACEDO-SILVA et al., 2014).
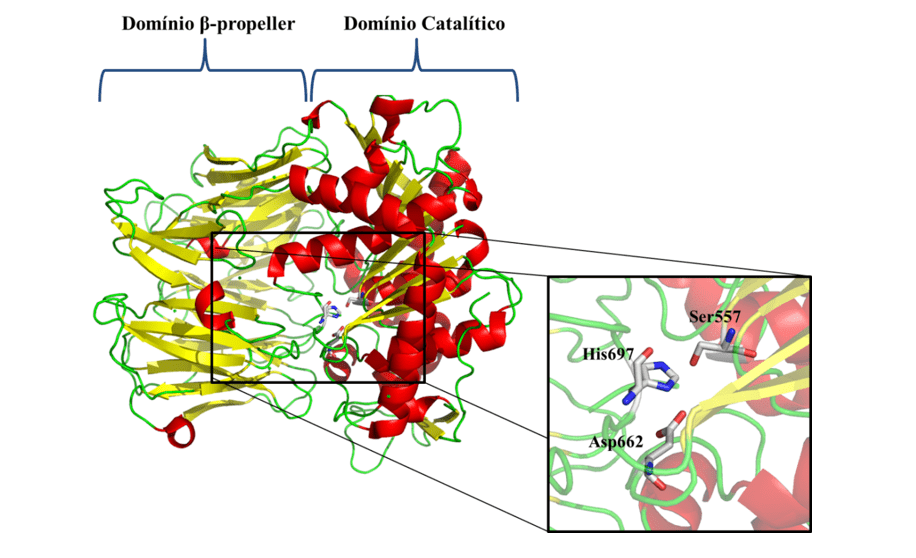
Tendo em vista as problemáticas apresentadas e a importância epidemiológica da leishmaniose, é notória a necessidade da investigação de novos alvos terapêuticos específicos contra a doença. Como um dos novos alvos terapêuticos, temos a Oligopeptidase B (OPB) (Figura 1), que é uma serino-protease, pertencente à subfamília S9A, tendo como característica uma tríade catalítica composta pelos resíduos de aminoácidos serina (Ser), ácido aspártico (Asp) e histidina (His) que se localizam entre os dois domínios. Essa enzima apresenta como característica a capacidade de clivar resíduos de estruturas proteicas. Além disso, é descrita na literatura como componente chave para o mecanismo de escape imune do parasita, clivando a enolase, uma proteína que irá opsonisar o protozoário, a fim de que ao ser reconhecido pelo macrófago o mesmo seja destruído. Porém ao passo que o parasita esteja no interior do macrófago, a OPB é super expressa, fazendo com que o parasita não seja reconhecido dentro da célula, deste modo o mesmo irá se multiplicar até lisar a mesma (SODERO et al., 2016; SWENERTON et al., 2011; OVCHINNIKOVA et al. 2018).
Figura 1: Estrutura tridimensional da OPB de L. major (código PDB 2XE4) (MCLUSKEY et al., 2010) mostrando os domínios catalítico e β-propeller. Estruturas secundárias como hélice-α, folhas-β e alças são mostradas em vermelho, amarelo e verde, respectivamente. No detalhe, são mostrados os resíduos (Ser557, Asp662 e His697) da tríade catalítica.
Por conseguinte, o estudo em questão teve por objetivo a investigação e caracterização das OPBs e de seus sítios ativos, das espécies L. brasiliensis, L, donovani, L. infantum, L. mexicana e L. panamensis. Por fim, esperou-se identificar possíveis semelhanças entre as proteínas, de modo que possibilite futuramente o desenvolvimento de uma nova terapia promissora com amplo espectro de ação sobre as enzimas de todas as espécies do estudo.
OBJETIVO GERAL
Tendo em vista a necessidade do desenvolvimento de novas entidades químicas para o tratamento contra as leishmanioses, este trabalho teve como objetivo principal construir e caracterizar a enzima oligopeptidase B de Leishmania spp. empregando as técnicas de estudos computacionais.
OBJETIVOS ESPECÍFICOS
- Realizar a construção e validação dos modelos das enzimas Oligopeptidase B das espécies de Leishmania;
- Realizar a caracterização das enzimas;
- Realizar estudos de simulação por modos normais.
MATERIAL E MÉTODOS
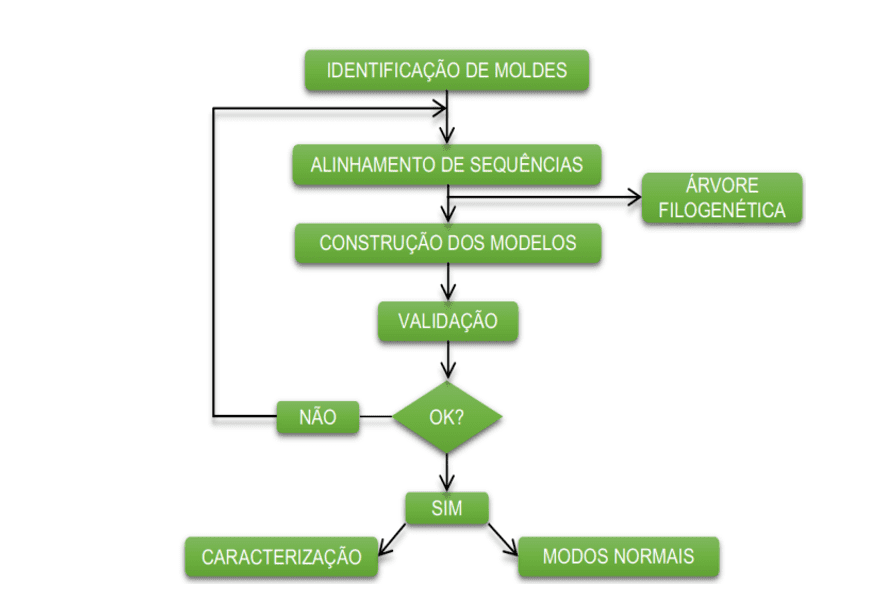
O esquema abaixo (esquema 1) mostra de forma simplificada as etapas empregadas durante o desenvolvimento deste trabalho.
Esquema 1: Esquema simplificado das etapas de material e métodos.
OBTENÇÃO DA ESTRUTURA PRIMÁRIA
As estruturas primárias das OPBs de Leishmanias foram obtidas do banco de dados Uniprot (The Universal Protein Resource) (WANG et al., 2012).
IDENTIFICAÇÃO DO MOLDE
Em seguida, foi realizada a busca por proteínas-molde a partir do alinhamento entre a sequência de aminoácidos da proteína-alvo e de sequências de aminoácidos de proteínas depositadas no Banco de Dados de Proteínas (PDB – Protein Data Bank) pelo servidor BLAST (Basic Local Alignment Search Tool) (ATLSCHUL et al., 1997) baseado na identidade entre as sequências de aminoácidos.
ALINHAMENTO DAS SEQUÊNCIAS
Uma vez definidos o molde a ser utilizado, a sequência do molde foi alinhada com as suas respectivas OPBs, no programa ClustalOmega (ClustaO) (SIEVERS et al., 2011).
CONSTRUÇÃO DO MODELO
Após a obtenção do alinhamento, essas informações foram utilizadas no programa MODELLER v9.20 para construção das estruturas 3D dos modelos (WEBB e SALI, 2016).
VALIDAÇÃO DOS MODELOS CONSTRUÍDOS
Para a validação dos modelos foi utilizado o gráfico de Ramachandran gerado pelo servidor PDBsum, Verify 3D e ProSA-web, todos obtidos no servidor Saves.
Na análise do gráfico de Ramachandran é permitida a visualização dos ângulos diédricos (phi (ψ) e psi (φ)) de resíduos de aminoácidos em estrutura proteica. O ângulo phi (ψ) é a resultante da ligação entre o grupo NH e carbono alfa, enquanto o ângulo psi (φ) é originado da ligação entre o carbono alfa e o grupo carbonila (RAMACHANDRAN et al., 1963).
O gráfico fornece uma maneira fácil de visualizar a distribuição dos ângulos de torção de uma estrutura de proteína. Além disso, fornece uma visão geral das regiões permitidas e não permitidas dos valores do ângulo de torção, servindo como um fator importante na avaliação da qualidade das estruturas 3D de proteínas. Com isso, define os resíduos que se encontram nas regiões energicamente mais favoráveis e desfavoráveis e orienta a avaliação da qualidade de modelos teóricos ou experimentais de proteínas. Este gráfico é divido em regiões, de modo que as regiões mais favoráveis estão em vermelho, regiões adicionais mais permissivas em marrom, regiões permissivas em amarelo e regiões não permissivas em branco. De acordo com essa validação para que um modelo predito seja considerado de excelente qualidade deve possuir mais de 90% dos resíduos de aminoácidos na região mais favorável (SANTOS-FILHO e ALENCASTRO, 2003).
Acrescido a isso, necessita ter a maioria de seus resíduos nas regiões mais favoráveis, bem como não possuir resíduos nas regiões não permissivas, exceto os aminoácidos glicina (Gly) e prolina (Pro) que são exceções para essa área. Esses dois resíduos apresentam variações na cadeia lateral que conferem maior rigidez, no caso da Pro, e maior flexibilidade, no caso da Gly, podendo assim assumir angulações não esperadas. Por esse motivo são aceitos nas regiões não permissivas do gráfico. As existências das regiões não permitidas se devem ao fato de existirem efeitos estéricos entre os resíduos (cadeias laterais) dos aminoácidos. (MORRIS et al., 1992).
O Verify 3D analisa a compatibilidade de um modelo atômico (3D) com sua própria sequência de aminoácidos (1D). Cada resíduo recebe uma classe estrutural baseada em sua localização e ambiente (alfa, beta, loop, polar, apolar, etc.). Logo após, um banco de dados gerado a partir de boas estruturas é usado para obter uma pontuação para cada um dos 20 aminoácidos nesta classe estrutural. O eixo vertical no gráfico representa a pontuação média do perfil 3D-1D para cada resíduo em uma janela deslizante de 21 resíduos e os resultados na forma de escores variam de -1 (má pontuação) a +1 (boa pontuação) (EISENBERG et al., 1997).
O ProSa-web calcula um escore de qualidade geral para uma estrutura de entrada específica. Se a pontuação desta estiver fora de uma característica de intervalo para proteínas nativas, a estrutura provavelmente contém erros. Um lote de pontuação de qualidade local aponta para partes problemáticas do modelo que também são destacadas em um visualizador de moléculas 3D para facilitar sua detecção (WIEDERSTEIN e SIPPL, 2007).
ANÁLISE DA ESTRUTURA SECUNDÁRIA
Depois da etapa de validação, as estruturas terciárias dos modelos foram comparadas com a distribuição da estrutura secundária predita pelo Quick2D, disponível no servidor Bioinformatics Toolkit (https://toolkit.tuebingen.mpg.de/), sendo assim escolhido os modelos (ALVA et al., 2016).
CARACTERIZAÇÃO DAS OLIGOPEPTIDASES (OPBS) DAS LEISHMANIAS
MAPA DO POTENCIAL ELETROSTÁTICO (MEP)
Para obtenção dos MEPs das superfícies e dos sítios de ligação das OPBs , foi utilizado uma extensão do programa PyMOL, o APB Tools (BAKER et al., 2001). Antes de serem analisadas no PyMOL, as enzimas foram preparadas no PDB2PQR Server (http://nbcr-222.ucsd.edu/pdb2pqr_2.0.0/) utilizando o parâmetro padrão do servidor (DOLINSKY et al., 2004).
CARACTERIZAÇÃO DOS SÍTIOS E SUBSÍTIOS DAS OPBS
Os modelos e o molde foram submetidos na plataforma proteins plus empregando a opção Dogsitescorer (VOLKAMER et al., 2012), em que foi feita a predição de cavidades nas estruturas 3D dos modelos. Essa gerou resultados referentes a possíveis sítios de ligação e subsítios das enzimas para a predição destas cavidades. O programa faz uso de uma malha tridimensional cuja aresta pode ser ajustada entre 0,2 Å e 1,0 Å, juntamente com um filtro gaussiano que é usado para identificar cavidades na superfície da proteína que sejam adequadas para acomodar átomos de ligantes. Além disso, o DoGSiteScorer também faz predição da drogabilidade para cada cavidade predita. Dessa forma, para cada interação entre a proteína e o possível fármaco é atribuída uma pontuação referente à drogabilidade da cavidade, denominada drugscore. Para a predição do valor do drugscore o programa faz uso de máquina de vetor suporte (SVM, do inglês, Support Vector Machine), em que são usados os seguintes descritores: volume, proporção de resíduos apolares e profundidade (VOLKAMER et al., 2012).
ÁRVORE FILOGENÉTICA
Por fim, foi analisado o grau de parentesco evolutivo entre as espécies de Leishmanias utilizando o programa MEGA (Molecular evulutionary genetics analysis), utilizando o método de Agrupamento de Vizinhos (Neighbor-joining methods), o qual permite a construção da árvore filogenética com finalidade de definir as proximidades evolutivas entre populações de sequências previamente definidas pelo usuário (KUMAR et al., 2004).
MODOS NORMAIS
Para a realização dos modos normais foram utilizados os arquivos gerados nas etapas de minimizações de energia feita pelo GROMACs versão 5.1.2. Foram realizadas as 4 primeiras etapas referentes ao processo de dinâmica molecular. A primeira foi a geração dos arquivos de topologia, com a adição de hidrogênios à proteína. Na segunda foi realizada a criação da caixa de água, sendo está etapa de suma importância para o cálculo da interação entre a proteína com o solvente. Na terceira foi feita a adição de íons para o estabelecimento de um sistema neutro. Por fim na quarta etapa foram realizadas as minimizações de energia, em que foi feito a inserção do campo de força AMBER99SB. A partir desse ponto foi realizado os modos normais dos modelos utilizando o servidor ANM (Anisotropic Network Model) (http://anm.csb.pitt.edu/) com objetivo de analisar o movimento das enzimas das espécies de Leishmania e, também, observar algumas características importantes para enzima, tais como, possíveis movimentos relacionados ao mecanismo de ação (EYAL et al., 2015).
RESULTADOS E DISCUSSÕES
SELEÇÃO DAS PROTEÍNAS MOLDES E ALINHAMENTO ENTRE AS SEQUÊNCIAS
Foram obtidas 100 estruturas primárias das OPBs de leishmania spp. utilizando o servidor UniProt, sendo 5 espécies de Leishmanias selecionadas. As sequências revisadas de aminoácidos selecionadas da enzima OPB foram L.brasiliensis, L. donovani, L. infantum, L. mexicana e L. panamensis sob os códigos A4H5Q8, C9EF60, A4HTZ8, E9AMS8 e A0A088RJA7, respectivamente Essas espécies foram selecionadas, devido apresentarem alta incidência na América do Sul e a sua resistência contra tratamento atual para leishmaniose (GHORBANI e FARHOUDI, 2018).
Para a construção dos modelos 3D, o programa BLAST foi utilizado para comparar as sequências de aminoácidos das sequências alvo com sequências de proteínas de estruturas tridimensionais experimentalmente elucidadas. Com base na identidade entre as sequências e o número de lacunas (gaps), foi selecionada a enzima OPB de L. major (código PDB 2XE4) como proteína molde (MCLUSKEY et al., 2010). A identidade entre as enzimas alvos e o seu respectivo molde apresentou valores entre 86 % a 96% (Tabela 1). O percentual de identidade entre duas sequências refere-se à presença do mesmo aminoácido na mesma posição entre as sequências alinhadas. Para a construção de um modelo de uma proteína com mais de 80 resíduos de aminoácidos, o percentual de identidade entre as estruturas primárias do molde e do modelo deve ser acima de 25%. Além disso, a porcentagem de lacunas (gaps) deve ser baixa de 20% para ser considerado um bom alinhamento (SANTOS-FILHO e ALENCASTRO, 2003). Dessa forma, a probabilidade de semelhança das estruturas tridimensionais das proteínas é alta.
Tabela 1: Percentual de identidade entre aos modelos das oligopeptidade B das espécies de Leishmania e o seu respectivo molde.
| Proteína molde (código PDB) | Modelos de OPB
(código uniprot) |
Identidade (%) | Gaps (%) |
| OPB
L. major (código PDB 2XE4) |
L.brasiliensis
(A4H5Q8) |
86 | 0 |
| L. donovani
(C9EF60) |
96 | 0 | |
| L. infantum
(A4HTZ8) |
96 | 0 | |
| L. Mexicana
(E9AMS8) |
90 | 0 | |
| L. panamensis
(A0A088RJA7) |
86 | 0 |
Fonte: Autoral.
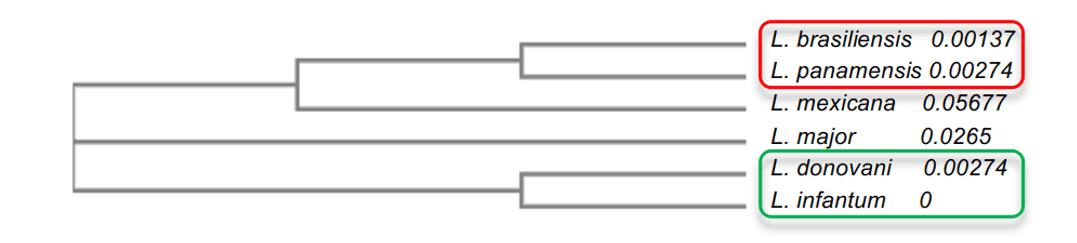
Em análise a árvore filogenética, foi possível explicar o grau de identidade entre as espécies de Leishmania. Foi observado que as espécies com maior grau de identidade, como a L. infantum e L. donovani, ambas com 96%, apresentaram uma proximidade entre si, além de estarem mais perto evolutivamente da L.major. Já as outras espécies apresentaram valores menores como a L.mexicana (90%), a qual ficou na mediana em relação aos outros modelos. Por fim os modelos de menor percentual de identidade, L. brasiliensis e L. panamensis, ambas com 86%, ficaram mais distante evolutivamente do seu molde, porém ficaram muito próximos entre si (Figura 2). Tal análise possibilitou a compreensão da diferença entre grau de identidade entre espécies com o molde e o entendimento de algumas características importantes da enzima entre as espécies.
Figura 2: Esquema da árvore filogenética das enzimas OPBs de Leishmania spp. Nesta representação é evidenciado os grupos de maior similaridade entre si, onde o grupo 1 está destacado em vermelho e o grupo 2 em verde.
MODELOS TRIDIMENSIONAIS DA ENZIMA DAS ESPÉCIES DE LEISHMANIA
Os modelos 3D das espécies foram construídos utilizando o programa Modeller (WEBB e SALI, 2016) a partir do alinhamento das estruturas primárias (figura 3 e 4).
Figura 3: Resultado do alinhamento entre as sequências primárias das OPBs selecionadas no UNIPROT (Parte 1/2).

Figura 4: Resultado do alinhamento entre as sequências primárias das OPBs selecionadas no UNIPROT (Parte 2/2).

Ao observar o resultado do alinhamento das estruturas (Figuras 3 e 4), notou-se um maior grau de semelhança, entre L. donovani e L. infantum (grupo 2) assim como, uma maior correspondência entre a L. brasiliensis e L. panamensis (grupo 1). Tais resultados reforçam o que foi apresentado pela árvore filogenética na Figura 2, onde é sugerido uma proximidade entre as espécies, L. donovani e L. infantum e outra proximidade entre as espécies de L. brasiliensis e L. panamensis. Em contrapartida, os dois grupos citados apresentaram uma maior distância evolutiva quando comparados entre si. Esse fato é notório quando feita uma comparação na análise do alinhamento, onde as estruturas primárias demonstraram uma maior diferença entre os resíduos, quando comparado os dois grupos. Além disso, o resultado do alinhamento da L. mexicana se mostrou bastante promissor o que se refere à sustentação desta discussão proposta, uma vez que, as diferenças observadas em comparação com as outras espécies, se mostraram equivalentes, ora ao primeiro grupo citado, ora ao segundo. Também foram observadas, em alguns trechos da sequência analisada, mutações pontuais que são características apenas da L. mexicana. Desse modo tal fato pode ser atestado devido a sua posição na mediana na árvore filogenética, em relação as demais espécies.
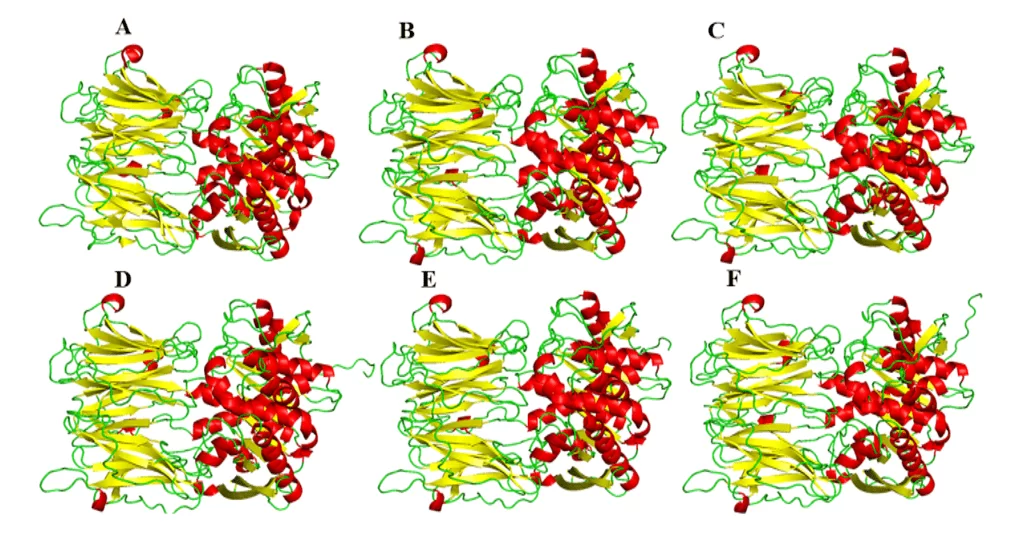
Como resultado da modelagem comparativa, foram obtidos os modelos do presente estudo, os quais, sugeriu uma grande semelhança visual na sua forma tridimensional em relação ao molde (Figura 5). Em relação a comparação entre os modelos, apresentaram quantidades diferentes nas estruturas de α-hélice e folha-β entre si. Essa diferença pode ser explicada pelas diferenças pontuais na composição dos resíduos da proteína, porém tais diferenças não se apresentaram em regiões importantes destas enzimas, como por exemplo, sítio de ligação. Com isso, essas diferenças não são tão importantes a ponto de modificar as estruturas ou o perfil de interação com um possível fármaco no sítio de ligação.
Figura 5: Modelos e molde das OPBs de leishmania spp., sendo evidenciado em vermelho as α-hélices, verde as alças e amarelo as folhas-β. Para OPB, (A) L.major (molde) e modelos: (B) L. brasiliensis, (C) L. donovani, (D) L. infantum, (E) L. mexicana e (F) L. panamensis.
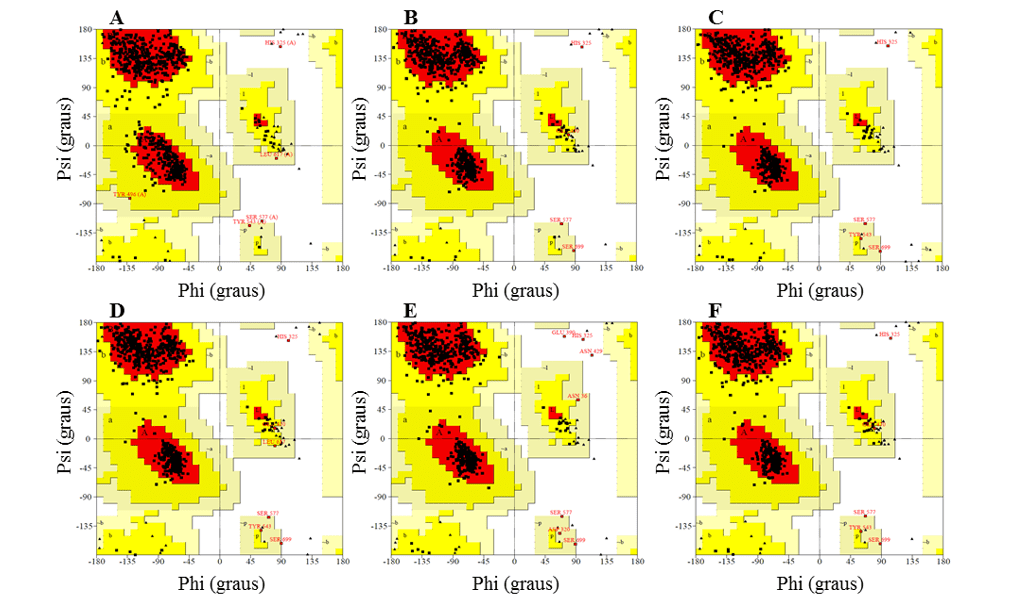
Na etapa de validação, foram analisados com base na análise do gráfico de Ramachandran (fornecido pelo servidor PDBsum), no score 3D-1D (fornecido pelo programa VERIFY-3D) e no score-Z (fornecido pelo servidor ProSA-web). Os valores obtidos para os modelos foram comparados àqueles obtidos para o molde.
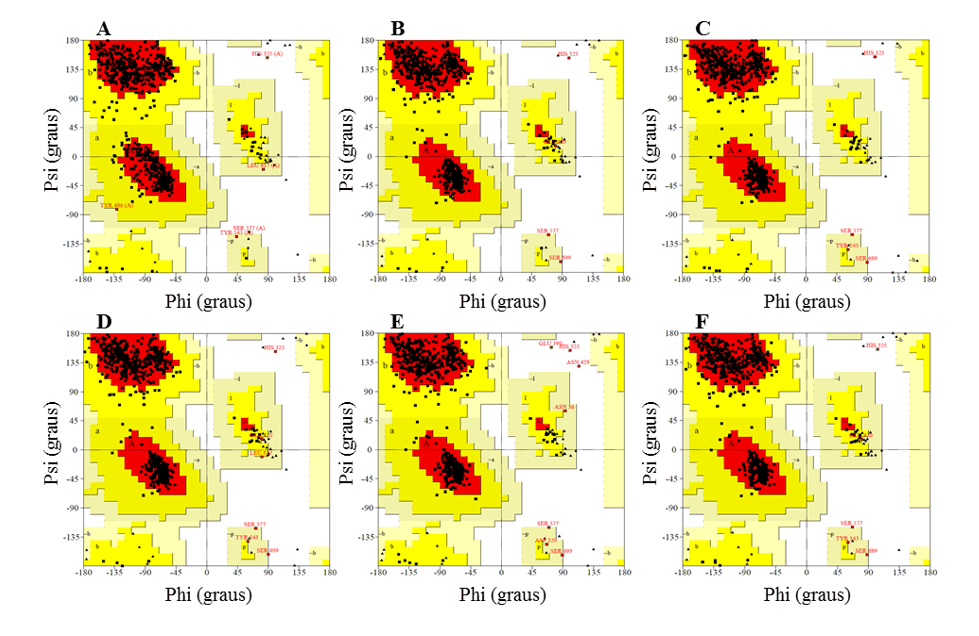
No gráfico de Ramachandran, os modelos apresentaram a maior parte dos resíduos nas regiões favoráveis, variando entre 91,2 e 92,3%, enquanto a percentagem de resíduos em regiões desfavoráveis foi no máximo de 0,5 %, sendo que os melhores modelos foram L.infantum e L.brasilienses. Esses modelos foram que apresentaram o maior número de resíduos em regiões favoráveis com 92,3% e 92,2% e a menor percentagem de resíduos em regiões desfavoráveis com 0,3 % e 0,2%, respectivamente (Figura 6).
Em todos os modelos foi possível observar que o resíduo Ser, que faz parte da tríade catalítica, estava nas regiões desfavoráveis. Contudo tal fato não afeta a validade dos modelos, visto que, ao comparar o molde apresentou o mesmo resultado. Sendo assim, esse resultado não configura uma baixa confiabilidade dos modelos.
Figura 6: Resultados dos gráficos de Ramachandran, obtidos pelo programa PROCHECK, das estruturas dos modelos de OPBs gerados, e do molde.
| Estruturas | % resíduos nas regiões | ||||
| Favoráveis | Permitidas | Desfavoráveis | |||
| OLIGOPEPTIDASE B | L. major (A)
(PDB 2XE4) |
90,2 | 9,5 | 0,3 | |
| L.brasilienses (B) | 92,2 | 7,7 | 0,2 | ||
| L. donovani (C) | 91,9 | 8,0 | 0,2 | ||
| L. infantum (D) | 92,3 | 7,3 | 0,3 | ||
| L. Mexicana (E) | 91,2 | 8,3 | 0,5 | ||
| L. panamensis (F) | 91,4 | 8,3 | 0,3 | ||
Fonte: Elaborado pelo autor com base nos resultados do PROCHECK.
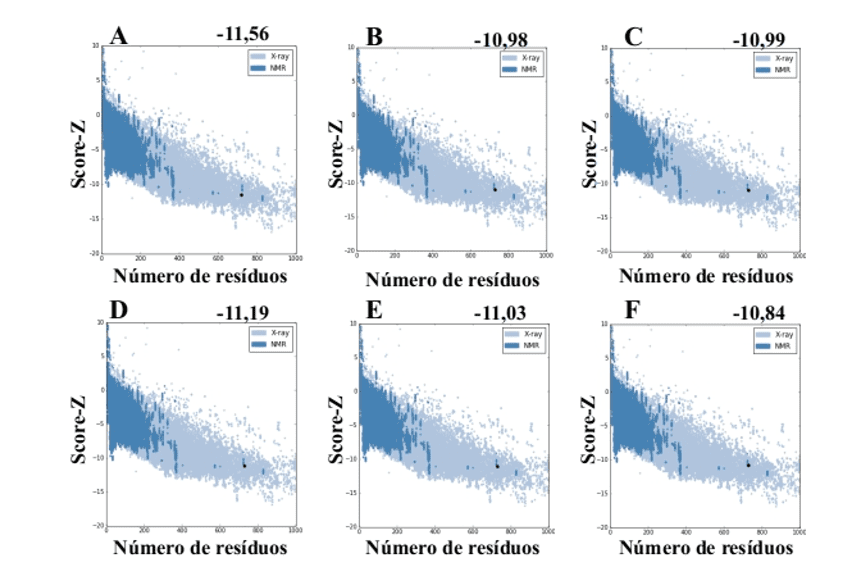
Ao utilizar o servidor ProSA-web a partir dos modelos gerados pelo MODELLER, esses apresentaram valores de score-Z -10,84 a -11,19 sendo esses valores compatíveis com estruturas do PDB (Figura 7).
Figura 7: Resultados de Z-score calculados no servidor ProSA-web das estruturas do molde (para comparação). (A) L .major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. A região em azul escuro indica o score das proteínas obtidas por RMN e em azul claro das proteínas obtidas por difração de raios-X.
Para avaliar a compatibilidade entre as estruturas 1D e 3D foi utilizado o programa Verify 3D. Os modelos obtidos apresentaram 95,28% a 98,34% dos aminoácidos com compatibilidade 3D-1D> 0,2, sendo que os modelos de L. donovani e L.brasilienses obtiveram melhores resultados. Segundo os parâmetros ideais do programa, a maior parte dos resíduos deve apresentar valores acima de zero, visto que valores abaixo de zero indicam regiões da molécula com problemas. O percentual dos aminoácidos com compatibilidade 3D-1D> 0,2 tem de ser acima de 80% (EISENBERG et al., 1997). Com isso, esses resultados indicam que os modelos apresentaram compatibilidade 1D-3D, sendo que os resíduos que apresentaram incompatibilidade não fazem parte do sítio ativo das enzimas (Tabela 2).
Tabela 2: Resultados do Verify 3D, evidenciando a percentagem de resíduos com score > 0,2.
| Estruturas | % de resíduos com score > 0,2 | |
| OLIGOPEPTIDASE B | L.major
(PDB 2XE4) |
93,20 |
| L.brasilienses | 95,62 | |
| L.donovani | 97,12 | |
| L.infantum | 94,93 | |
| L.mexicana | 95,62 | |
| L.panamensis | 94,93 |
Fonte: Autoral.
VALIDAÇÃO DA TRÍADE CATALÍTICA DA ENZIMA DOS MODELOS
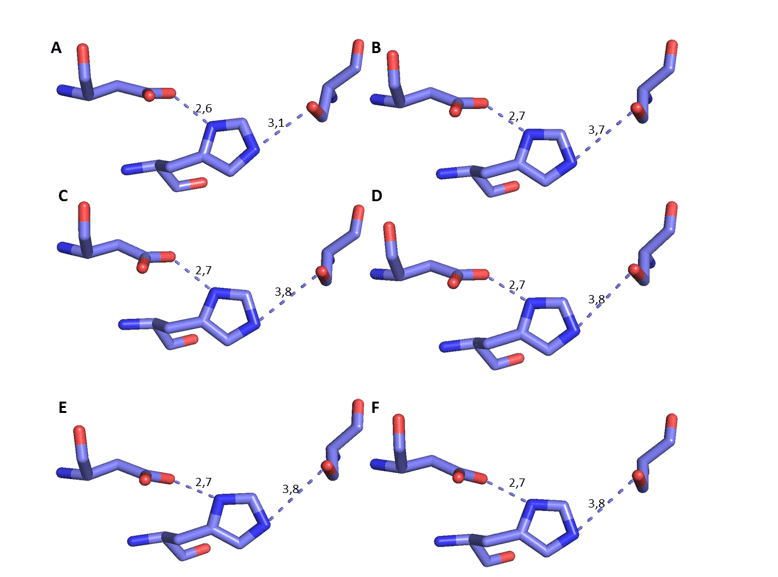
Devido ao mecanismo da OPB, é essencial analisar a distância e orientação entre os resíduos de aminoácidos a tríade catalítica (Ser, Asp e His) dos modelos gerados a fim de aumentar a confiabilidade dos modelos.
É possível observar a comparação desses resíduos específicos dos modelos de OPB com o molde em questão na figura 17. Além disso, foi possível medir a distância entre os resíduos da tríade catalítica, baseado no mecanismo de interação entre o sítio e o substrato, tendo a His como referência (DEREWENDA et al. , 1994). Portanto, é necessário que haja uma distância específica para ocorrer o mecanismo de ação. Essa distância deve ser de cerca de 3,5Å entre o oxigênio da His e o nitrogênio da Ser, além da distância de aproximadamente 2,6Å entre o oxigênio do aspartato e o outro nitrogênio da His, conforme está descrito na literatura (DEREWENDA et al. , 1994). Nessa análise foi possível constatar que a distância em questão, sofreu uma pequena variação, estando ainda de acordo com o que já foi descrito. Essa distância é importante, pois as serino-proteases necessitam da His para que a clivagem do substrato ocorra (HEDSTROM, 2002) (Figura 8). Baseado nesses resultados confiáveis foi dado prosseguimento em outras etapas de caracterização dos modelos.
Figura 8: Representação 3D da tríade catalítica das OPBs, respectivamente, Ser, His e Asp. A cor azul escuro representa o átomo de nitrogênio, a vermelha o oxigênio e a lilás o carbono. Para OPB, (A) L.major (molde)e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis.
CARACTERIZAÇÃO DA OLIGOPEPTIDASES DA LEISHMANIAS
PREDIÇÃO ESTRUTURA SECUNDÁRIA
A partir da predição da estrutura secundária das enzimas OPBs, foram reveladas o número e posição das estruturas secundárias dos modelos. O PSISPRED apresentou entre 15 e 16 estruturas α-hélices e todas apresentaram 38 pares de β- folhas (Tabela 3). A partir dos resultados gerados pelo PSIPRED foi realizada uma comparação visual dessa estrutura secundária predita com as estruturas tridimensionais dos 5 modelos, utilizando o programa Pymol. Os modelos de L.brasilienses, e L. panamensis foram os que apresentaram melhor similaridade, em relação às estruturas preditas secundária e terciária (2D-3D), apresentando 15 α-hélices e 38 de β-folhas. Das 15 α-hélices apresentadas na estrutura tridimensional do modelo 8 das α-hélices estavam na mesma posição prevista pelo PSIPRED. Essas hélices correspondem à sequência de aminoácidos: 59 a 74, 78 a 93, 535 a 540, 545 a 563, 631 a 640, 703 a 721 e 727 a 730.
Na análise de modo geral, não houve uma diferença exacerbada nas quantidades de α-hélices e folhas β entre as predições e os modelos obtidos (Tabela 3).
Tabela 3: Comparação entre as estruturas secundárias previstas pelo PSIPRED e o encontrado através do Pymol.
| Modelos | α-Hélices | Folhas β | PSIPRED
α-Hélices |
PSIPRED
Folhas β |
| L.brasiliensis | 15 | 38 | 11 | 36 |
| L. donovani | 16 | 38 | 11 | 36 |
| L. infantum | 16 | 38 | 11 | 36 |
| L. mexicana | 15 | 38 | 10 | 36 |
| L. panamensis | 15 | 38 | 11 | 36 |
Fonte: Autoral.
Ademais, foi possível observar que a quantidade de 15 α-Hélices foi igual para L.brasilienses, L. Mexicana e L. panamensis. Assim como L.infantum e L.donovani obtiveram o número de 16 α-Hélice. Segundo árvore filogenética, esses dois grupos citados estão no mesmo nó interno e são consideradas como monofilético (Figura 2).
Em seguida, foi realizado o RMSD (root-mean-square deviation) entre os modelos e o molde. Os valores se mostraram promissores, como é possível notar na tabela 4, pois os RMSDs não ultrapassaram o valor de 0,19 Å. Tal achado pode ser justificado pelo alto grau de identidade entre o molde e os respectivos modelos. De maneira geral, é esperado que proteínas com identidade superior a 30%, tenham excelente sobreposição das cadeias principais, deste modo, é obtido um RMSD da ordem de 2Å (BENNER et al., 1997 ; CHOTHIA et al., 1986).
Tabela 4: RMSDs das OPBs geradas pelo Modeller, tendo com orientação os carbonos alfa do molde de OPB de L. major.
| Molde | Modelos | RMSD (Å) |
| L. major
(2XE4) |
L.brasiliensis | 0,15 |
| L. donovani | 0,15 | |
| L. infantum | 0,16 | |
| L. Mexicana | 0,19 | |
| L. panamensis | 0,14 |
Fonte: Autoral.
MAPA DO POTENCIAL ELETROSTÁTICO MOLECULAR (MEP) DA SUPERFÍCIE DAS ENZIMAS E DOS RECEPTIVOS SÍTIOS
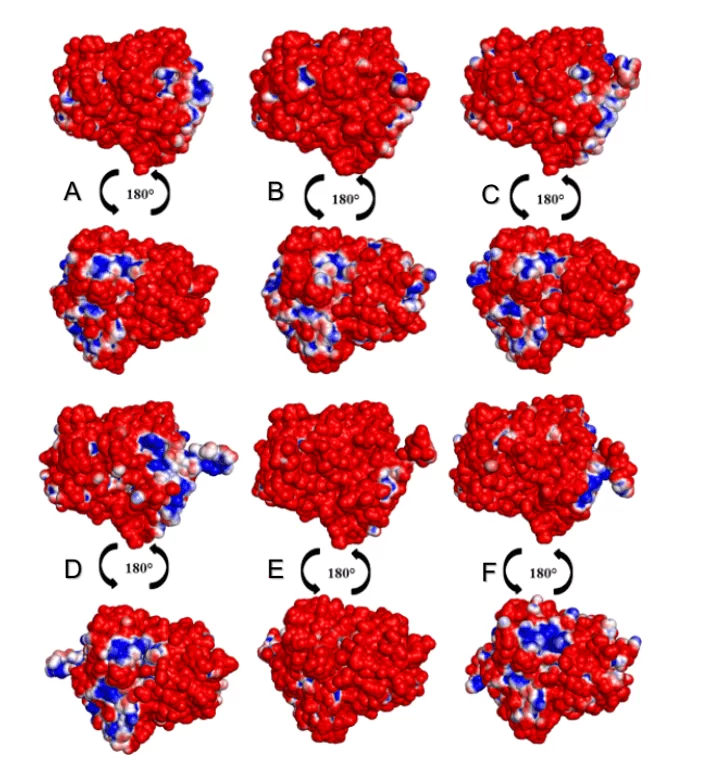
Na análise dos MEPs das superfícies das OPBs, foi possível observar que todas as espécies de leishmania apresentaram uma maior porcentagem de regiões negativas do que positivas conforme é possível observar na figura 15. As espécies L. donovani e L. infantum (grupo verde) sugeriam uma área negativa (na cor azul) na mesma região. Já as espécies L.brasilienses e L. panamensis (grupo vermelho) apresentaram uma região negativa semelhante, em um padrão colorimétrico semelhante. Ambos os resultados podem ser justificados pelo fato de que as espécies comparadas entre si são pertencentes ao mesmo monofilético (Figura 2).
Figura 9: Mapa do potencial eletrostático dos modelos 3D de OPBs de Leishmania spp e o do seu molde. Para OPB, (A) L.major (molde) e modelos: (B) L. brasilienses (C) L. donovani, (D) L. infantum, (E) L. mexicana (F) L. panamensis. Na coloração azul apresenta a região positiva e na coloração vermelha, a região negativa.
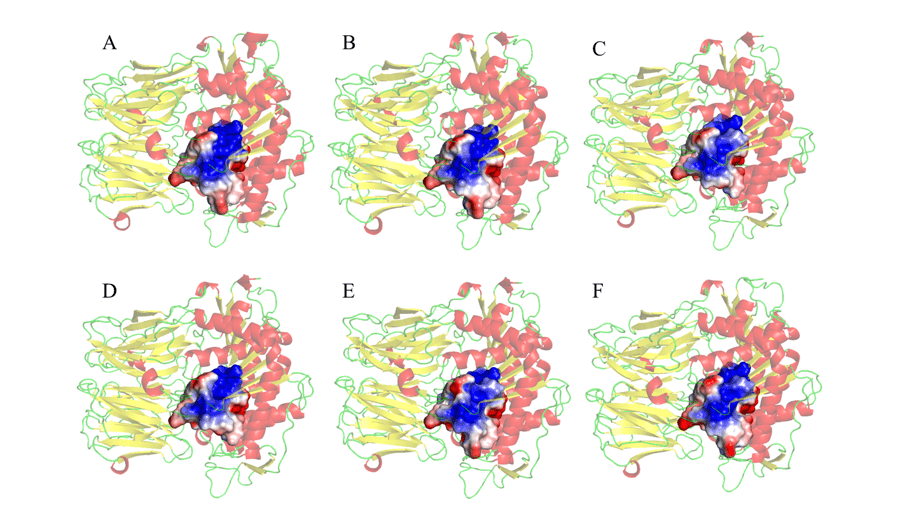
Por fim, também foi realizado o MEP dos resíduos a um raio de distância de 5 Å em torno da tríade catalítica, conforme é evidenciado na figura 10. A escolha de se observar o MEP da região em torno da tríade catalítica se deu pelo fato dessa ser a segunda região de interação entre o a enzima e o substrato, no qual haverá acomodação (ANDERSSON et al., 2010). Desse modo tendo em vista um dos objetivos do trabalho, foi possível constatar semelhanças e correspondências significativas entre o molde e os modelos testados. Tais resultados, como indicado pela figura 10, revelaram principalmente uma maior porção eletropositiva (em azul) na região central dos sítios de ligação. Foram observadas regiões negativas (em vermelho) em áreas periféricas dos MEPs estudados. Esses resultados são promissores, pois se constatou que tais regiões se mostram plenamente semelhantes em todos os modelos, podendo auxiliar no desenvolvimento de um fármaco que tenha a capacidade de atuar de maneira específica em todos os modelos estudados.
Figura 10: Representação do mapa de potencial da superfície eletrostática dos resíduos de aminoácidos, 5Å em torno da tríade catalítica que compõem o sítio ativo da enzima. (A) L.major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. Em vermelho estão as α-hélices, verde as alças e amarelo as folhas-β. Na cor dourada o sitio de ligação. Na coloração azul apresenta a região positiva e na coloração vermelha, a região negativa.
DOGSITESCORER
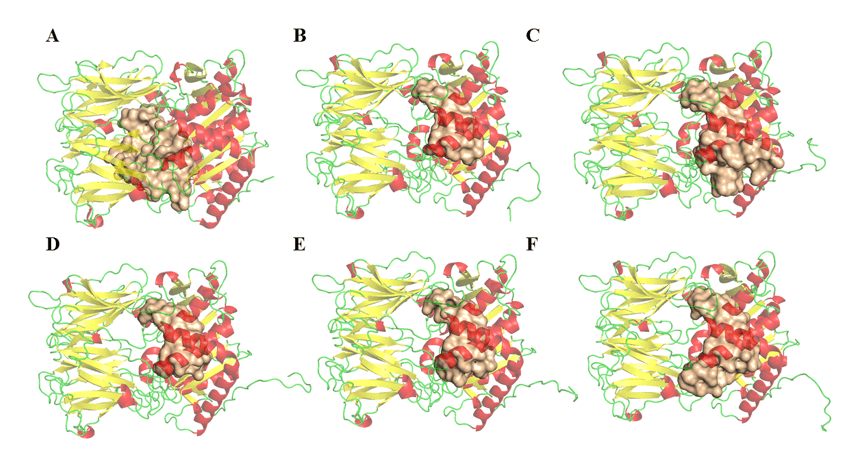
A determinação dos parâmetros de volume, área e profundidade dos possíveis sítios de ligação das enzimas OPBs das espécies de Leishmania foi realizada no programa DoGSiteScorer (http://poseview.zbh.uni-hamburg.de/) da ProteinsPlus – Structure-Based Modeling Support Server (VOLKAMER et al., 2012). O programa aponta três sítios de ligação como é descrito na literatura. Na figura 11, foi possível observar a comparação entre a possível região do sitio ativo e suas diferenças. Essas variáveis entre as OPBs podem estar relacionadas aos resíduos que não são descritos como importantes para sua inibição, mas foi considerado durante essa análise. Com isso, esse resultado não descarta a possibilidade da antipaína ou de qualquer outra molécula ter um amplo espectro de inibição sobre a enzima, devido o fato de que os resultados referentes ao drugscore foram bem similares e positivos em todas as regiões. Pode ser observada na tabela 5, uma relativa discrepância entre os resultados de volume e área das cavidades encontradas nos modelos. Tal resultado não sugere um viés de dúvida sobre o potencial inibitório de um único fármaco nas respectivas enzimas. Porém, atenta para o fato de que algumas proteínas apresentam cavidades maiores que outras. Contudo estas mesmas cavidades tem pontos em comum, e são estes os resíduos que compõem o sítio catalítico, como pode ser observado na figura 11 e podem ser explorados.
Tabela 5: Valores referentes as possíveis regiões de ligação das OPBs (obtidos pelo servidor).
| Estruturas | DogSiteScoore | |||
| Volume | Área | Drug Score | ||
| OLIGOPEPTIDASE B | L. major
(PDB 2XE4) |
1690,62 | 1818,41 | 0,80 |
| L.brasiliensis | 1527,84 | 1766,63 | 0,80 | |
| L. donovani | 1074,57 | 1428,55 | 0,79 | |
| L. infantum | 1309,97 | 1572,19 | 0,80 | |
| L. mexicana | 800,92 | 799,38 | 0,85 | |
| L. panamensis | 971,96 | 1083,40 | 0,81 | |
Fonte: Autoral.
Figura 11: Estruturas de OPBs e possíveis regiões de ligação (obtidos pelo servidor DogSite). (A) L .major (molde)e modelos: (B) L. brasilienses, (C), L. donovani, (D) L. infantum, (E) L. mexicana e (F) L. panamensis. Em vermelho estão as α-hélices, verde as alças e amarelo as folhas-β. Na cor dourada o sitio de ligação.
MODOS NORMAIS
Depois da caracterização da enzima nos aspectos estruturais, superficiais e dos sítios de ligação, foram realizados os modos normais para as enzimas de cada espécie com finalidade dos seus respectivos movimentos.
Após o relaxamento e as minimizações de energia realizadas pela dinâmica molecular no GROMACS, foram submetidas estas estruturas as análises por modos normais, a fim de se observar possíveis movimentos compatíveis.
Foi possível observar em todos os modelos estudados, um movimento expressivo de uma α hélice específica, e ao analisar tal região em sua composição de aminoácidos é possível notar que a mesma se mostra altamente conservada. Desse modo o padrão de movimento se repetiu em todos os modelos em questão. Tal região sugeriu um movimento linear se afastando do centro em direção a periferia, expondo a tríade catalítica. Isso pode ser um indicativo do movimento realizado pela proteína para a acomodação do substrato.
Ao observar a figura 12 e 13, pode-se notar o mesmo padrão de cores em todos os modelos do estudo. Esses dados representam a capacidade que dada região tem de movimentar em um determinado sentido. As cores azuis representam regiões mais invariáveis durante a simulação por modos normais, com isso, nota-se um padrão de rigidez nas proteínas correspondendo principalmente às folhas-β do domínio β-propeller e algumas α-hélices que compõem o domínio catalítico. As cores próximas ao verde representam regiões intermediárias em relação à capacidade e amplitude de movimento. Com base nessa informação é possível perceber essa coloração presente em loops nas extremidades dos modelos assim como em algumas α-hélices do domínio catalítico. Por fim, há aquelas regiões que apresentaram uma coloração laranja/vermelha, no qual é a representação de um enorme potencial de movimentação. Desse modo, pode-se constatar que existem pequenas regiões de loops nas extremidades dos domínios catalíticos com este potencial de movimentação, assim como uma α-hélice situada na porção do domínio catalítico, mais especificamente em frente à tríade catalítica das OPBs, no qual obteve a movimentação mais expressiva do estudo.
Todos os modelos obtiveram a mesma configuração de movimento, onde uma α-hélice (em cor laranja) demonstrou ser a região com maior capacidade de movimentação. Dessa forma, as OPBs, de L. brasiliensis, L. donovani, L. infantum, L. mexicana e L. panamensis em suas maiores amplitudes de movimentos obtiveram, respectivamente, 9.8 Å, 11.3Å, 11.8Å, 11.9Å e 11.6Å (figura 12 e 13).
Figura 12: Resultado da análise por modos normais em representação pelo programa pymol, na imagem é evidenciado os movimentos dos modelos, em suas formas relaxadas (direita) em comparação com o movimento de maior amplitude (esquerda). Também é representado as regiões mais rígidas (Azul escuro), regiões com pouca movimentação (Azul claro), regiões intermediárias (Verde), regiões com bom potencial de movimentação (Laranja) e regiões extremamente maleáveis (vermelho) (Parte1/2).
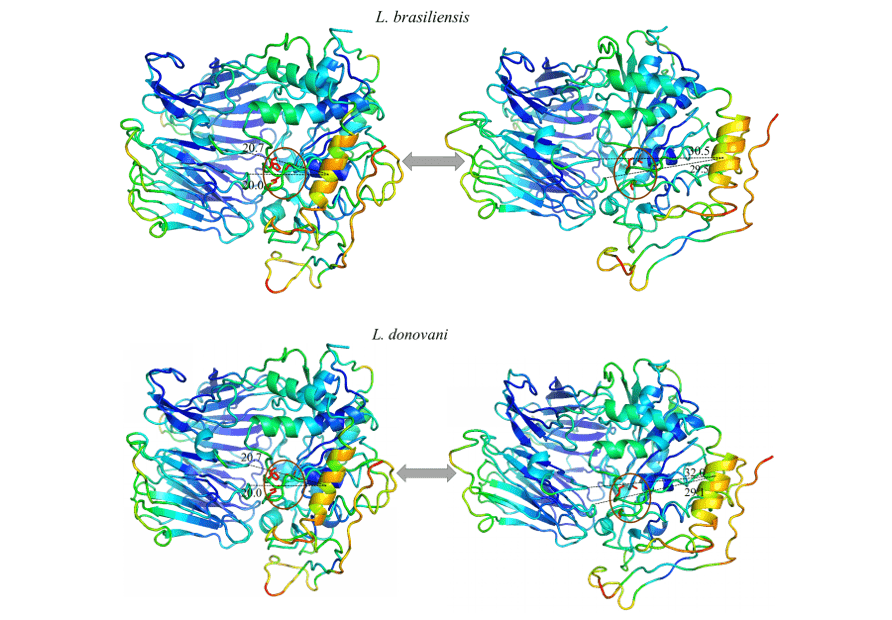
Figura 13: Resultado da análise por modos normais em representação pelo programa pymol, na imagem é evidenciado os movimentos dos modelos, em suas formas relaxadas (direita) em comparação com o movimento de maior amplitude (esquerda). Também é representado as regiões mais rígidas (Azul escuro), regiões com pouca movimentação (Azul claro), regiões intermediárias (Verde), regiões com bom potencial de movimentação (Laranja) e regiões extremamente maleáveis (vermelho).(Parte 2/2)
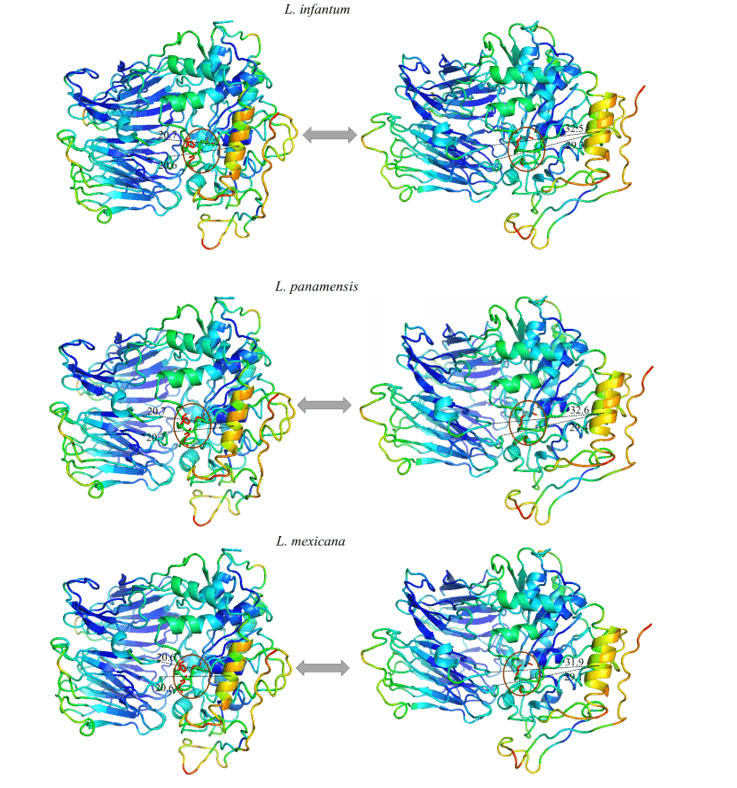
Em vista disso, os estudos por modos normais endossam um possível mecanismo das OPBs ainda não descrito para as espécies de Leishmania spp. até o presente estudo.
CONCLUSÃO
Nesse estudo foram obtidos os modelos tridimensionais da enzima OPB de L.brasiliensis, L. donovani, L. infantum, L. mexicana e L. panamensis. A validação dos modelos apresentou resultados confiáveis para todos os modelos tridimensionais obtidos. Na caracterização da enzima, o mapa de potencial eletrostático superficial demonstrou que a maior parte dos resíduos apresentou carga negativa. Já na caracterização da região em torno da tríade catalítica, demonstrou semelhança entre o volume, área e correspondência entre resíduos positivos e negativos. Por conseguinte, foi possível constatar que os resultados da análise por modos normais sugeriam, um movimento expressivo em uma de α hélice específica, ocorrendo um afastamento linear desta, do centro em direção a periferia, expondo assim, a tríade catalítica. A descrição destas movimentações realizadas por esta enzima é de grande importância para auxiliar o entendimento do funcionamento da mesma.
Por fim, os resultados do presente trabalho podem agregar conhecimento para a comunidade científica, trazendo elucidações e novos questionamentos relacionados ao tema, servindo de base para eventuais estudos na área da saúde.
REFERÊNCIAS
A. Benner S, Cannarozzi G, Gerloff D, Turcotte M, Chelvanayagam G. Bona Fide Predictions of Protein Secondary Structure Using Transparent Analyses of Multiple Sequence Alignments. Chem Rev. 1997;97(8):2725-2844. doi:10.1021/cr940469a.
Alva, V., Nam, S. Z., Söding, J., & Lupas, A. N. (2016). The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Research, 44(W1), W410–W415. doi.org/10.1093/nar/gkw348.
Alvarenga DG, Escalda PMF, da Costa ASV, Monreal MTFD. Leishmaniose visceral: Estudo retrospectivo de fatores associados à letalidade. Rev Soc Bras Med Trop. 2010;43(2):194-197.
Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389‐3402. doi:10.1093/nar/25.17.3389.
Andersson CD, Chen BY, Linusson A. Mapping of ligand-binding cavities in proteins [published correction appears in Proteins. 2011 Apr;79(4):1363]. Proteins. 2010;78(6):1408–1422. doi:10.1002/prot.22655.
Bailey F, Mondragon-Shem K, Hotez P, et al. A new perspective on cutaneous leishmaniasis-Implications for global prevalence and burden of disease estimates. PLoS Negl Trop Dis. 2017;11(8):e0005739. Published 2017 Aug 10. doi:10.1371/journal.pntd.0005739.
Carmo RF, Luz ZMP da, Bevilacqua PD. Percepções da população e de profissionais de saúde sobre a leishmaniose visceral. Cien Saude Colet. 2016;21(2):621-628. doi:10.1590/1413-81232015212.10422015.
Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5(4):823‐826.
Derewenda ZS, Derewenda U, Kobos PM. (His)Cε-H···O=C< Hydrogen Bond in the Active Sites of Serine Hydrolases. J Mol Biol. 1994;241(1):83-93. doi:10.1006/JMBI.1994.1475.
Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32(Web Server issue):W665–W667. doi:10.1093/nar/gkh381.
Eisenberg, D., Lüthy, R., & Bowie, J. U. (1997). [20] VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods in Enzymology, 277, 396–404. https://doi.org/10.1016/S0076-6879(97)77022-8.
Eyal E, Lum G, Bahar I. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics. 2015;31(9):1487–1489. doi:10.1093/bioinformatics/btu847.
Ghorbani Masoud, Farhoudi Ramin. Leishmaniasis in humans: drug or vaccine therapy? Drug Des Devel Ther. 2018;12:25-40. doi:10.2147/DDDT.S146521.
Hedstrom L. Serine Protease Mechanism and Specificity. Chem Rev. 2002;102(12):4501-4524. doi:10.1021/cr000033x.
Katsila T, Spyroulias GA, Patrinos GP, Matsoukas MT. Computational approaches in target identification and drug discovery. Comput Struct Biotechnol J. 2016;14:177–184. Published 2016 May 7. doi:10.1016/j.csbj.2016.04.004.
Kumar S, Tamura K, Nei M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5(2):150-163.
Macedo-Silva RM, dos Santos C de LP, Diniz VA, De Carvalho JJ, Guerra C, Côrte-Real S. Peripheral blood fibrocytes: New information to explain the dynamics of Leishmania infection. Mem Inst Oswaldo Cruz. 2014;109(1):61-69. doi:10.1590/0074-0276130247
Machado P de A, Carneiro MPD, Sousa-Batista A de J, et al. Leishmanicidal therapy targeted to parasite proteases. Life Sci. 2019;219:163-181. doi:10.1016/J.LFS.2019.01.015.
Morris, A. L., MacArthur, M. W., Hutchinson, E. G., & Thornton, J. M. (1992). Stereochemical quality of protein structure coordinates. Proteins: Structure, Function, and Bioinformatics, 12(4), 345–364. https://doi.org/10.1002/prot.340120407.
Ovchinnikova M V., Mikhailova AG, Karlinsky DM, Gorlenko VA, Rumsh LD. Reversible cyclic thermal inactivation of oligopeptidase B from Serratia proteamaculans. Acta Naturae. 2018;10(2):65-70.
Ramachandran, G. N., Ramakrishnan, C., & Sasisekharan, V. (1963). Stereochemistry of polypeptide chain configurations. Journal of Molecular Biology, 7(1), 95–99. https://doi.org/10.1016/S0022-2836(63)80023-6.
Santos Filho, O. A., & Alencastro, R. B. de. (2003). Modelagem de proteínas por homologia. Química Nova, 26(2), 253–259. https://doi.org/10.1590/S0100-40422003000200019
Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. Published 2011 Oct 11. doi:10.1038/msb.2011.75.
SODERO ACR, DOS SANTOS ACGO, MELLO JFRE, et al. Oligopeptidase B and B2: comparative modelling and virtual screening as searching tools for new antileishmanial compounds. Parasitology. 2017;144(4):536-545. doi:10.1017/s0031182016002237.
Swenerton RK, Zhang S, Sajid M, et al. The oligopeptidase B of Leishmania regulates parasite enolase and immune evasion. J Biol Chem. 2011;286(1):429-440. doi:10.1074/jbc.M110.138313.
Volkamer, A., Kuhn, D., Rippmann, F., & Rarey, M. (2012). Dogsitescorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics, 28(15), 2074–2075. https://doi.org/10.1093/bioinformatics/bts310.
Wang Q, Arighi CN, King BL, et al. Community annotation and bioinformatics workforce development in concert–Little Skate Genome Annotation Workshops and Jamborees. Database (Oxford). 2012;2012:bar064. Published 2012 Mar 20. doi:10.1093/database/bar064
Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics. 2016;54:5.6.1–5.6.37. Published 2016 Jun 20. doi:10.1002/cpbi.3.
Wiederstein, M., & Sippl, M. J. (2007). ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research, 35(SUPPL.2), 407–410. https://doi.org/10.1093/nar/gkm290.
WHO. Integrating Neglected Tropical Diseases into Global Health and Development: Fourth WHO Report on Neglected Tropical Diseases.; 2017. http://apps.who.int/iris/bitstream/10665/255011/1/9789241565448-eng.pdf?ua=1.
WHO. (2019). Leishmanioses – Informe Epidemiológico das Américas No 7 – Março, 2019. Retrieved from http://iris.paho.org/xmlui/bitstream/handle/123456789/50505/ 2019-cde-leish-informe-epi-das-americas.pdf?sequence=2&isAllowed=y.
ANEXO – FIGURAS E TABELAS EM INGLÊS
Esquema 1: Esquema simplificado das etapas de material e métodos.
Tabela 1: Percentual de identidade entre aos modelos das oligopeptidade B das espécies de Leishmania e o seu respectivo molde.
| Mold protein (code PDB) | Models OPB
(código uniprot) |
Identity (%) | Gaps (%) |
| OPB
L. major (code PDB 2XE4) |
L.brasiliensis
(A4H5Q8) |
86 | 0 |
| L. donovani
(C9EF60) |
96 | 0 | |
| L. infantum
(A4HTZ8) |
96 | 0 | |
| L. Mexicana
(E9AMS8) |
90 | 0 | |
| L. panamensis
(A0A088RJA7) |
86 | 0 |
Fonte: Autoral.
Figura 6: Resultados dos gráficos de Ramachandran, obtidos pelo programa PROCHECK, das estruturas dos modelos de OPBs gerados, e do molde.
| Structures | % waste in the regions | ||||
| Favorable | Allowed | Unfavorable | |||
| OLIGOPEPTIDASE B | L. major (A)
(PDB 2XE4) |
90,2 | 9,5 | 0,3 | |
| L.brasilienses (B) | 92,2 | 7,7 | 0,2 | ||
| L. donovani (C) | 91,9 | 8,0 | 0,2 | ||
| L. infantum (D) | 92,3 | 7,3 | 0,3 | ||
| L. Mexicana (E) | 91,2 | 8,3 | 0,5 | ||
| L. panamensis (F) | 91,4 | 8,3 | 0,3 | ||
Fonte: Elaborado pelo autor com base nos resultados do PROCHECK.
Figura 7: Resultados de Z-score calculados no servidor ProSA-web das estruturas do molde (para comparação). (A) L .major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. A região em azul escuro indica o score das proteínas obtidas por RMN e em azul claro das proteínas obtidas por difração de raios-X
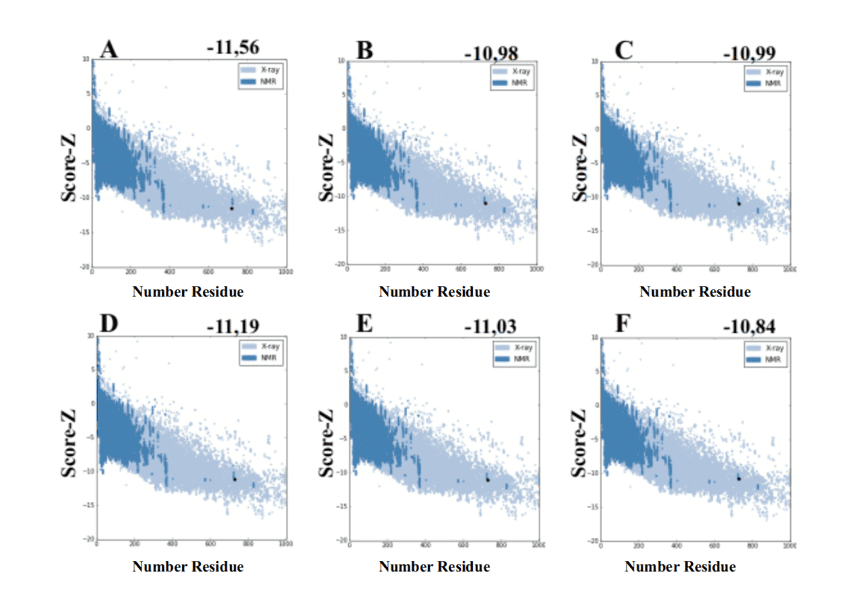
Tabela 2: Resultados do Verify 3D, evidenciando a percentagem de resíduos com score > 0,2.
| Structures | % residue with score > 0,2 | |
| OLIGOPEPTIDASE B | L.major
(PDB 2XE4) |
93,20 |
| L.brasilienses | 95,62 | |
| L.donovani | 97,12 | |
| L.infantum | 94,93 | |
| L.mexicana | 95,62 | |
| L.panamensis | 94,93 |
Fonte: Autoral.
Tabela 3: Comparação entre as estruturas secundárias previstas pelo PSIPRED e o encontrado através do Pymol.
| Models | α-Hélix | Sheet β | PSIPRED
α-Hélix |
PSIPRED
Sheet β |
| L.brasiliensis | 15 | 38 | 11 | 36 |
| L. donovani | 16 | 38 | 11 | 36 |
| L. infantum | 16 | 38 | 11 | 36 |
| L. mexicana | 15 | 38 | 10 | 36 |
| L. panamensis | 15 | 38 | 11 | 36 |
Fonte: Autoral.
Tabela 4: RMSDs das OPBs geradas pelo Modeller, tendo com orientação os carbonos alfa do molde de OPB de L. major.
| Mold | Models | RMSD (Å) |
| L. major
(2XE4) |
L.brasiliensis | 0,15 |
| L. donovani | 0,15 | |
| L. infantum | 0,16 | |
| L. mexicana | 0,19 | |
| L. panamensis | 0,14 |
Fonte: Autoral.
Tabela 5: Valores referentes as possíveis regiões de ligação das OPBs (obtidos pelo servidor).
| Structures | DogSiteScoore | |||
| Volume | Area | Drug Score | ||
| OP OLIGOPEPTIDASE B | L. major
(PDB 2XE4) |
1690,62 | 1818,41 | 0,80 |
| L.brasiliensis | 1527,84 | 1766,63 | 0,80 | |
| L. donovani | 1074,57 | 1428,55 | 0,79 | |
| L. infantum | 1309,97 | 1572,19 | 0,80 | |
| L. mexicana | 800,92 | 799,38 | 0,85 | |
| L. panamensis | 971,96 | 1083,40 | 0,81 | |
Fonte: Autoral.
[1] Biomédico e mestrando em Bioquímica Médica.
[2] Mestre em Ciências Farmacêuticas e Farmacêutica pela UFRJ.
[3] Doutora em Química, Mestre em Química Orgânica e Farmacêutica industrial.
Enviado: Maio, 2020.
Aprovado: Maio, 2020.