ARTICLE ORIGINAL
RIBEIRO, Fernando de Sá [1], JESUS, Jéssica Barbosa de [2], SOUZA, Alessandra Mendonça Teles de [3]
RIBEIRO, Fernando de Sá. JESUS, Jéssica Barbosa de. SOUZA, Alessandra Mendonça Teles de. Analyse et caractérisation d’une cible thérapeutique prometteuse identifiée dans Leishmania spp. Revista Científica Multidisciplinar Núcleo do Conhecimento. Ano 05, Ed. 05, Vol. 09, pp. 99-132. Maio de 2020. ISSN: 2448-0959, Lien d’accès: https://www.nucleodoconhecimento.com.br/sante/target-therapeutique
RÉSUMÉ
Leishmaniose est une maladie négligée causée par les protozoaires du genre Leishmania spp., qui touche environ 1,6 million d’individus chaque année et 500 000 se présentent sous la forme viscérale. Au Brésil, il y a environ 30 000 nouveaux cas chaque année. En outre, le pays est responsable de 90% des cas signalés de leishmaniose viscérale, et c’est une forme plus grave de la maladie. Allié à ces faits, le traitement actuel est inefficace, contribuant à l’établissement de souches résistantes. Actuellement, le traitement a plusieurs effets secondaires et des dommages permanents à la santé des patients, ce fait a contribué à la recherche de nouveaux médicaments contre la leishmaniose. L’enzyme oligopeptidase B (OPB) a été étudiée comme cible thérapeutique possible dans le développement d’agents antiparasitaires. Ainsi, l’objectif de ce travail est de construire le modèle tridimensionnel de l’enzyme Oligopeptidase B de différentes espèces de Leishmania spp. et les comparer les uns aux autres. À cette fin, la méthode de modélisation comparative a été utilisée. Dans cette méthode, les modèles de l’espèce L. brasiliensis, L. donovani, L. infantum, L. mexicana et L. panamensis ont été construits à l’aide du programme MODELLER. Une fois les modèles prêts, le processus de validation a été effectué et par la suite caractérisé, ce qui a été possible de vérifier un degré prometteur de similitude entre les modèles. Enfin, ces modèles ont été soumis à la méthode d’analyse par des modes normaux, qui ont obtenu un modèle de mouvement similaire, il a donc été possible de vérifier un mouvement dans une région spécifique d’une alpha-hélice, conduisant par conséquent à la triade de l’enzyme étant exposée, ce qui peut être indicatif d’un mécanisme d’action. Enfin, il devrait utiliser les modèles construits pour aider au développement d’une nouvelle thérapie prometteuse pour le traitement de la leishmaniose.
Mots-clés: Leishmaniasis, oligopeptidase B, modélisation moléculaire, modes normaux.
INTRODUCTION
Leishmaniose, causée par la leishmaniose spp., est une maladie caractérisée par plusieurs types de manifestations, de doux, dans lequel il ya des rapports de petites lésions que même sans régression due de traitement aux plus graves tels que la leishmaniose viscérale (VL). Au Brésil, la forme la plus grave de cette maladie présente des données alarmantes par rapport à d’autres pays, faisant du pays le plus grand détenteur de cas de VL dans toute l’Amérique (ALVARENGA, 2010; QUI, 2019).
Cette maladie appartient au groupe de maladies négligées, qui font partie de toutes les maladies qui touchent principalement les pays sous-développés et les régions les plus pauvres, de sorte qu’elle ne suscite pas d’intérêt pour le développement de médicaments. Par conséquent, il est nécessaire que des techniques efficaces et peu coûteuses survivent pour surmonter ce manque d’incitations financières pour l’étude de cette maladie. Ainsi, des méthodes de calcul peuvent être utilisées afin de réduire le temps dans le développement d’une nouvelle thérapie prometteuse et, par conséquent, le coût par rapport aux méthodes plus traditionnelles pour le développement de médicaments (BAILEY et al., 2017; QUI, 2017).
Malgré le peu d’investissements dans ce domaine, il existe un traitement pour la maladie comme les pentas antimonial valentes (médicament de premier choix) ou l’amphotericine B (médicament de deuxième choix). Cependant, ces traitements ont plusieurs inconvénients tels que le taux de résistance élevé et la grande variété d’effets secondaires, allant du mal de mer aux problèmes possibles causés à la troisième (3e) paire du nerf crânien, conduisant à des difficultés motrices (MACEDO-SILVA et al., 2014).
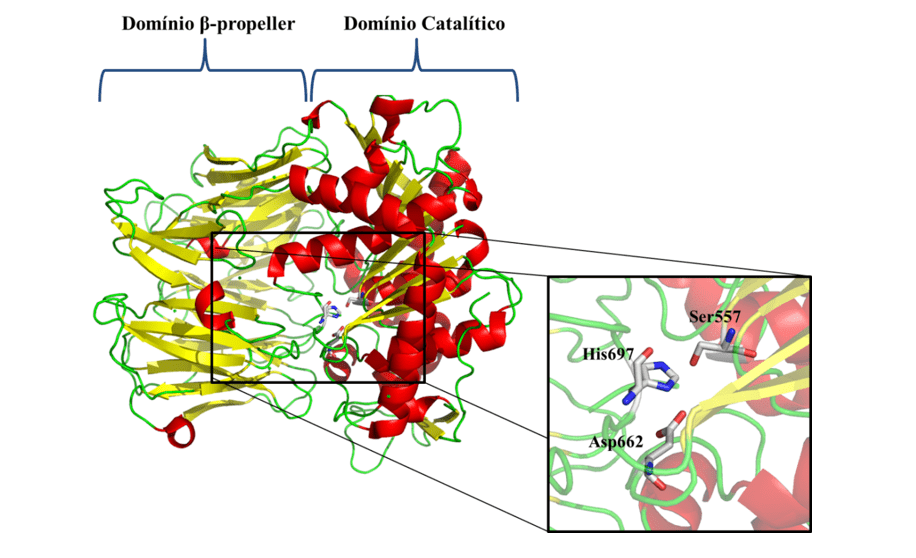
Compte tenu des problèmes présentés et de l’importance épidémiologique de la leishmaniose, la nécessité d’enquêter sur de nouvelles cibles thérapeutiques spécifiques contre la maladie est notoire. Comme l’une des nouvelles cibles thérapeutiques, nous avons Oligopeptidase B (OPB) (figure 1), qui est une protéase sérine, appartenant à la sous-famille S9A, ayant comme caractéristique une triade catalytique composée des résidus d’acides aminés sérine (Ser), acide asaptique (Asp) et histidine (son) qui sont situés entre les deux domaines. Cette enzyme a comme caractéristique la capacité de fendre les résidus des structures protéiques. En outre, il est décrit dans la littérature comme un composant clé pour le mécanisme d’évacuation immunitaire du parasite, clivant la olase, une protéine qui opsonisar le protozoaire, de sorte que lorsqu’il est reconnu par le macrophage, il est détruit. Cependant, alors que le parasite est à l’intérieur du macrophage, l’OPB est super exprimé, ce qui provoque le parasite de ne pas être reconnu dans la cellule, de sorte qu’il se multipliera jusqu’à ce qu’il soit lissé (SODERO et al., 2016; SWENERTON et coll., 2011; OVCHINNIKOVA et coll. 2018).
Figure 1 : Structure tridimensionnelle de L. major OPB (code PDB 2XE4) (MCLUSKEY et coll., 2010) montrant les domaines catalytiques et à hélice. Les structures secondaires telles que l’hélice, les feuilles et les poignées sont indiquées en rouge, jaune et vert, respectivement. En particulier, les résidus (Ser557, Asp662 et His697) de la triade catalytique sont montrés.
Par conséquent, l’étude en question visait à étudier et caractériser les BPO et leurs sites actifs, de l’espèce L. brasiliensis, L, donovani, L. infantum, L. mexicana et L. panamensis. Enfin, on s’attendait à ce qu’il identifie les similitudes possibles entre les protéines, afin qu’il permette le développement futur d’une nouvelle thérapie prometteuse avec un large éventail d’actions sur les enzymes de toutes les espèces dans l’étude.
OBJECTIF GÉNÉRAL
Compte tenu de la nécessité de développer de nouvelles entités chimiques pour le traitement contre la leishmaniose, ce travail avait pour objectif principal de construire et de caractériser l’enzyme oligopeptidase B de Leishmania spp. en utilisant des techniques d’étude computationnelle.
OBJECTIFS SPÉCIFIQUES
- Construire et valider les modèles d’enzymes oligopeptidase B d’espèces leishmania;
- Effectuer la caractérisation des enzymes;
- Effectuez des études de simulation par des modes normaux.
MATÉRIEL ET MÉTHODES
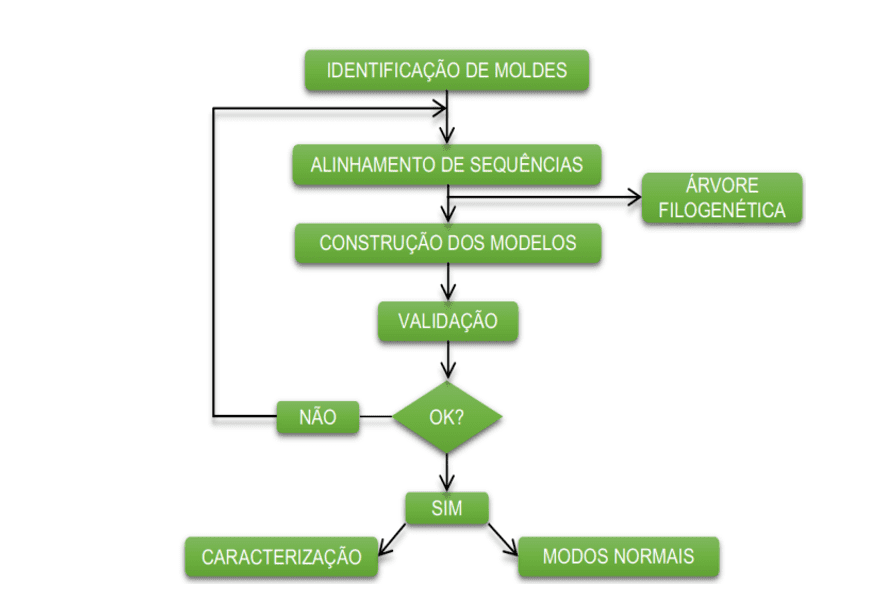
Le diagramme ci-dessous (diagramme 1) montre de manière simplifiée les étapes utilisées lors du développement de ce travail.
Schéma 1 : Schéma simplifié des étapes et des méthodes matérielles.
OBTENIR LA STRUCTURE PRIMAIRE
Les structures primaires des BPO leishmanias ont été obtenues à partir de la base de données Uniprot (The Universal Protein Resource) (WANG et coll., 2012).
ID DE MOULE
Ensuite, la recherche de protéines de moisissure a été effectuée à partir de l’alignement entre la séquence d’acides aminés de la protéine cible et les séquences d’acides aminés des protéines déposées dans la Banque de données protéinées (PDB) par le serveur BLAST (Basic Local Alignment Search Tool) (ATLSCHUL et al., 1997) basé sur l’identité entre les séquences d’acides aminés.
ALIGNEMENT DES SÉQUENCES
Une fois que le moule à utiliser a été défini, la séquence de moule a été alignée avec leurs OPB respectifs, dans le programme ClustalOmega (ClustaO) (SIEVERS et al., 2011).
CONSTRUCTION DE MODÈLE
Après avoir obtenu l’alignement, cette information a été utilisée dans le programme MODELLER v9.20 pour construire les structures 3D des modèles (WEBB et SALI, 2016).
VALIDATION DES MODÈLES CONSTRUITS
Pour la validation des modèles, le graphique Ramachandran généré par le serveur PDBsum, Verify 3D et ProSA-web a été utilisé, le tout obtenu à partir du serveur Saves.
Dans l’analyse du graphique Ramachandran, il est permis de visualiser les angles dihedral (phi (phi) et psi (en) des résidus d’acide aminé dans la structure des protéines. L’angle de phi est le résultat du lien entre le groupe NH et le carbone alpha, tandis que l’angle de psi (ô) provient du lien entre le carbone alpha et le groupe carbonyle (RAMACHANDRAN et al., 1963).
Le graphique fournit un moyen facile de visualiser la distribution des angles torm d’une structure protéique. En outre, il fournit un aperçu des régions autorisées et non autorisées des valeurs d’angle de torm, servant de facteur important dans l’évaluation de la qualité des structures de protéines 3D. Avec cela, il définit les résidus qui se trouvent dans les régions qui sont énergiquement plus favorables et défavorables et guide l’évaluation de la qualité des modèles théoriques ou expérimentaux de protéines. Ce graphique est divisé en régions, de sorte que les régions les plus favorables sont en rouge, d’autres régions plus permissives dans les régions brunes et permissives dans les régions jaunes et non-permissives en blanc. Selon cette validation d’un modèle prévu pour être considéré comme d’excellente qualité, il doit avoir plus de 90% des résidus d’acide aminé dans la région la plus favorable (SANTOS-FILHO et ALENCASTRO, 2003).
En outre, il a besoin d’avoir la majorité de ses résidus dans les régions les plus favorables, ainsi que de ne pas avoir de résidus dans les régions non-permissives, à l’exception des acides aminés glycine (Gly) et proline (Pro) qui sont des exceptions pour ce domaine. Ces deux résidus présentent des variations dans la chaîne latérale qui confèrent une plus grande rigidité, dans le cas de Pro, et une plus grande flexibilité, dans le cas de Gly, ce qui peut supposer des angulations inattendues. Pour cette raison, ils sont acceptés dans les régions non-permissives du graphique. L’existence des régions non autorisées est due au fait qu’il y a des effets stériques parmi les résidus (chaînes latérales) d’acides aminés. (MORRIS et al., 1992).
Verify 3D analyse la compatibilité d’un modèle atomique (3D) avec sa propre séquence d’acide aminé (1D). Chaque résidu reçoit une classe structurelle en fonction de son emplacement et de son environnement (alpha, bêta, boucle, polaire, apolaire, etc.). Peu de temps après, une base de données générée à partir de bonnes structures est utilisée pour obtenir un score pour chacun des 20 acides aminés de cette classe structurelle. L’axe vertical du graphique représente la note moyenne du profil 3D-1D pour chaque résidu résiduel dans une fenêtre coulissante de 21 résidus et les résultats sous forme de scores varient de -1 (mauvais score) à 1 (bon score) (EISENBERG et al., 1997).
ProSa-web calcule un score de qualité global pour une structure d’entrée spécifique. Si le score de ceci est en dehors d’une gamme caractéristique pour les protéines indigènes, la structure contient probablement des erreurs. Beaucoup de scores de qualité locale indiquent des parties problématiques du modèle qui sont également mises en évidence dans un visualiseur de molécule 3D pour une détection facile (WIEDERSTEIN et SIPPL, 2007).
ANALYSE DE LA STRUCTURE SECONDAIRE
Après l’étape de validation, les structures tertiaires des modèles ont été comparées à la distribution de la structure secondaire prédite par Quick2D, disponible sur le serveur Bioinformatics Toolkit (https://toolkit.tuebingen.mpg.de/), choisissant ainsi les modèles (ALVA et al., 2016).
CARACTÉRISATION DES OLIGOPEPTIDASES (OPBS) DES LEISHMANIES
CARTE ÉLECTROSTATIQUE POTENTIELLE (MEP)
Pour obtenir les eurodéputés des surfaces et des sites de connexion des BPO, une extension du programme PyMOL, des outils apb (BAKER et coll., 2001) a été utilisé. Avant d’être analysées dans PyMOL, les enzymes ont été préparées dans le serveur PDB2PQR (http://nbcr-222.ucsd.edu/pdb2pqr_2.0.0/) en utilisant le paramètre standard du serveur (DOLINSKY et al., 2004).
CARACTÉRISATION DES SITES ET SOUS-SITES OPBS
Les modèles et le moule ont été soumis dans les protéines plus la plate-forme en utilisant l’option Dogsitescorer (VOLKAMER et al., 2012), dans lequel les cavités ont été prévues dans les structures 3D des modèles. Ceci a généré des résultats concernant les sites de liaison possibles et les sous-sols d’enzyme pour la prédiction de ces cavités. Le programme utilise un maillage tridimensionnel dont le bord peut être ajusté entre 0,2 et 1,0 , ainsi qu’un filtre gaussien qui est utilisé pour identifier les cavités à la surface de la protéine qui conviennent pour accueillir les atomes ligand. En outre, DoGSiteScorer prédit également la pharmacovisibilité pour chaque cavité prévue. Ainsi, pour chaque interaction entre la protéine et le médicament possible, un score se référant à la drugability de cavité, appelé drugscore, est attribué. Pour prédire la valeur de drugscore, le programme utilise une machine vectorielle de soutien (SVM), dans laquelle les descripteurs suivants sont utilisés : volume, proportion des résidus nonpolaires et de la profondeur (VOLKAMER et al., 2012).
ARBRE PHYLOGENÉTIQUE
Enfin, le degré de parenté évolutive entre les espèces de Leishmanias a été analysé à l’aide du programme MEGA (analyse génétique épileptionnaire moléculaire), à l’aide des méthodes d’adhésion des voisins, qui permet la construction de l’arbre phylogénétique afin de définir les proximités évolutives entre les populations de séquences précédemment définies par l’utilisateur (KUMAR et al., 2004).
MODES NORMAUX
Pour exécuter les modes normaux, les fichiers générés dans les stades de l’énergie minimizaçs fabriqués par la version 5.1.2 des GROMACs ont été utilisés. Les 4 premières étapes liées au processus de dynamique moléculaire ont été effectuées. Le premier a été la génération de fichiers topologiques, avec l’ajout d’hydrogènes à la protéine. Dans la seconde, la boîte à eau a été créée, et c’est une étape très importante pour le calcul de l’interaction entre la protéine et le solvant. Dans le troisième, des ions ont été ajoutés pour établir un système neutre. Enfin, dans la quatrième étape, des minimisations énergétiques ont été effectuées, dans lesquelles le champ de force AMBER99SB a été inséré. À partir de ce moment, les modes normaux des modèles ont été exécutés à l’aide du serveur ANM (Anisotropic Network Model) (http://anm.csb.pitt.edu/) afin d’analyser le mouvement des enzymes des espèces de Leishmania et aussi d’observer certaines caractéristiques importantes pour l’enzyme, telles que les mouvements possibles liés au mécanisme d’action (EYAL et al., 2015).
RÉSULTATS ET DISCUSSIONS
SÉLECTION DES PROTÉINES DE MOISISSURE ET ALIGNEMENT ENTRE LES SÉQUENCES
Nous avons obtenu 100 structures primaires des BPO de la leishmania spp. à l’aide du serveur UniProt, avec 5 espèces de Leishmanias sélectionnées. Les séquences d’acides aminés révisées sélectionnées à partir de l’enzyme OPB étaient L.brasiliensis, L. donovani, L. infantum, L. mexicana et L. panamensis sous les codes A4H5Q8, C9EF60, A4HTZ8, E9AMS8 et A0A088RJA7, respectivement Ces espèces ont été sélectionnées, en raison de leur incidence élevée en Amérique du Sud et de leur résistance contre le traitement actuel de la leishmaniose (GHORBANI et FARHOUDI, 2018).
Pour la construction de modèles 3D, le programme BLAST a été utilisé pour comparer les séquences d’acides aminés des séquences cibles avec des séquences protéiques de structures tridimensionnelles expérimentalement élucidées. Basé sur l’identité entre les séquences et le nombre de lacunes, l’ENZYME OPB de L. major (code PDB 2XE4) a été sélectionnée comme protéine de moule (MCLUSKEY et al., 2010). L’identité entre les enzymes cibles et leur moule respectif présentait des valeurs comprises entre 86 % et 96 % (tableau 1). Le pourcentage d’identité entre deux séquences se réfère à la présence d’un même acide aminé dans la même position entre les séquences alignées. Pour la construction d’un modèle de protéine avec plus de 80 résidus d’acides aminés, le pourcentage d’identité entre les structures primaires du moule et le modèle devrait être supérieur à 25%. En outre, le pourcentage d’écarts doit être faible de 20 % pour être considéré comme un bon alignement (SANTOS-FILHO et ALENCASTRO, 2003). Ainsi, la probabilité de similitude des structures tridimensionnelles des protéines est élevée.
Tableau 1 : Pourcentage d’identité entre les modèles d’oligopeptité B des espèces de Leishmania et leur moule respectif.
| Proteína molde (código PDB) | Modelos de OPB
(código uniprot) |
Identidade (%) | Gaps (%) |
| OPB
L. major (código PDB 2XE4) |
L.brasiliensis
(A4H5Q8) |
86 | 0 |
| L. donovani
(C9EF60) |
96 | 0 | |
| L. infantum
(A4HTZ8) |
96 | 0 | |
| L. Mexicana
(E9AMS8) |
90 | 0 | |
| L. panamensis
(A0A088RJA7) |
86 | 0 |
Source: Auteur.
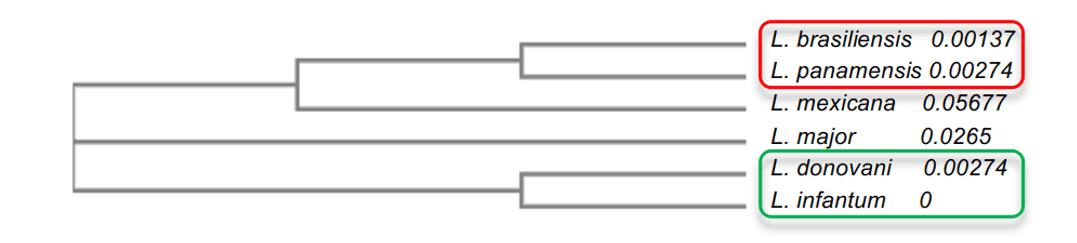
Dans l’analyse phylogénétique de l’arbre, il a été possible d’expliquer le degré d’identité parmi les espèces de leishmaniose. On a observé que les espèces ayant un degré d’identité plus élevé, comme L. infantum et L. donovani, toutes deux avec 96 %, se présentaient à proximité l’une de l’autre, en plus d’être plus proches de L.major. Les autres espèces présentaient des valeurs inférieures comme L.mexicana (90 %), qui était dans la médiane par rapport aux autres modèles. Enfin, les modèles ayant le plus faible pourcentage d’identité, L. brasiliensis et L. panamensis, tous deux avec 86%, étaient plus éloignés de leur moule, mais étaient très proches les uns des autres (figure 2). Cette analyse a permis de comprendre la différence entre le degré d’identité entre les espèces et la compréhension de certaines caractéristiques importantes de l’enzyme chez l’espèce.
Figure 2 : Schéma d’arbre phylogénétique des enzymes leishmania spp. OPBs. Dans cette représentation, les groupes ayant la plus grande similitude les uns avec les autres sont mis en évidence, où le groupe 1 est mis en évidence en rouge et dans le groupe 2 en vert.
MODÈLES TRIDIMENSIONNELS DE L’ENZYME DES ESPÈCES DE LEISHMANIA
Les modèles 3D de l’espèce ont été construits à l’aide du programme Modeller (WEBB et SALI, 2016) à partir de l’alignement des structures primaires (figure 3 et 4).
Figure 3 : Résultat de l’alignement entre les séquences primaires des BPO sélectionnées dans le PNUPROP (partie 1/2).

Figure 4 : Résultat de l’alignement entre les séquences primaires des BPO sélectionnées dans le PNUPROP (partie 2/2).

En observant le résultat de l’alignement des structures (figures 3 et 4), un degré plus élevé de similitude a été observé entre L. donovani et L. infantum (groupe 2) ainsi qu’une plus grande correspondance entre L. brasiliensis et L. panamensis (groupe 1). Ces résultats renforcent ce qui a été présenté par l’arbre phylogénétique à la figure 2, où il est suggéré une proximité entre l’espèce, L. donovani et L. infantum et une autre proximité entre les espèces de L. brasiliensis et L. panamensis. D’autre part, les deux groupes mentionnés ont présenté une plus grande distance évolutive par rapport à l’autre. Ce fait est notoire lorsqu’une comparaison est faite dans l’analyse d’alignement, où les structures primaires ont montré une plus grande différence entre les résidus, par rapport aux deux groupes. En outre, le résultat de l’alignement de L. mexicana s’est avéré très prometteur, ce qui fait référence à l’appui de cette discussion proposée, puisque les différences observées par rapport aux autres espèces étaient équivalentes, parfois au premier groupe mentionné, parfois au second. Dans certaines parties de la séquence analysée, des mutations spécifiques qui ne sont caractéristiques que de L. mexicana ont également été observées dans certaines parties de la séquence analysée. Ainsi, ce fait peut être attesté en raison de sa position dans la médiane dans l’arbre phylogénétique, par rapport aux autres espèces.
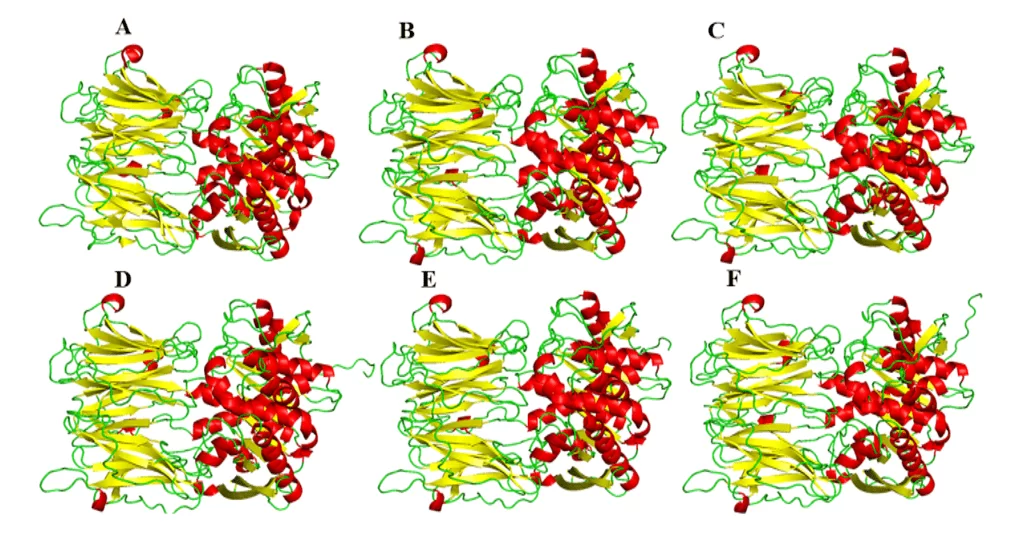
À la suite de la modélisation comparative, les modèles de la présente étude ont été obtenus, ce qui a suggéré une grande similitude visuelle dans sa forme tridimensionnelle par rapport au moule (figure 5). En ce qui concerne la comparaison entre les modèles, ils présentaient des quantités différentes dans les structures de l’hélice et des feuilles entre eux. Cette différence peut s’expliquer par les différences spécifiques dans la composition des résidus de protéines, mais de telles différences ne se présentaient pas dans les régions importantes de ces enzymes, telles que le site de liaison. Par conséquent, ces différences ne sont pas si importantes que de modifier les structures ou le profil de l’interaction avec un médicament possible sur le site de liaison.
Figure 5 : Modèles et moules des BPO de la leishmania spp., étant démontrés en rouge les hélices, verdir les poignées et jaune les feuilles. Para OPB, (A) L.major (molde) e modelos: (B) L. brasiliensis, (C) L. donovani, (D) L. infantum, (E) L. mexicana e (F) L. panamensis.
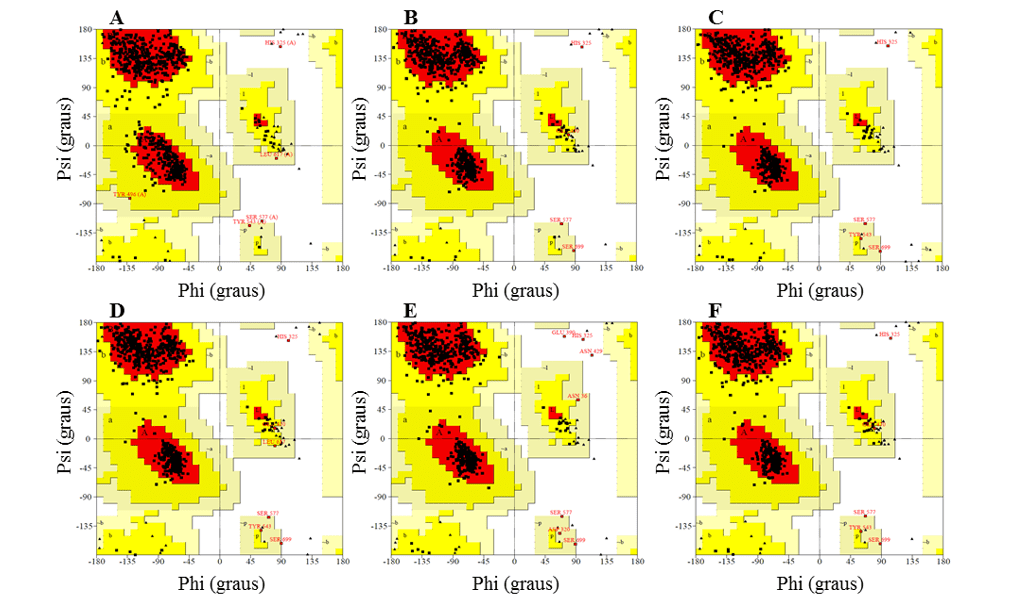
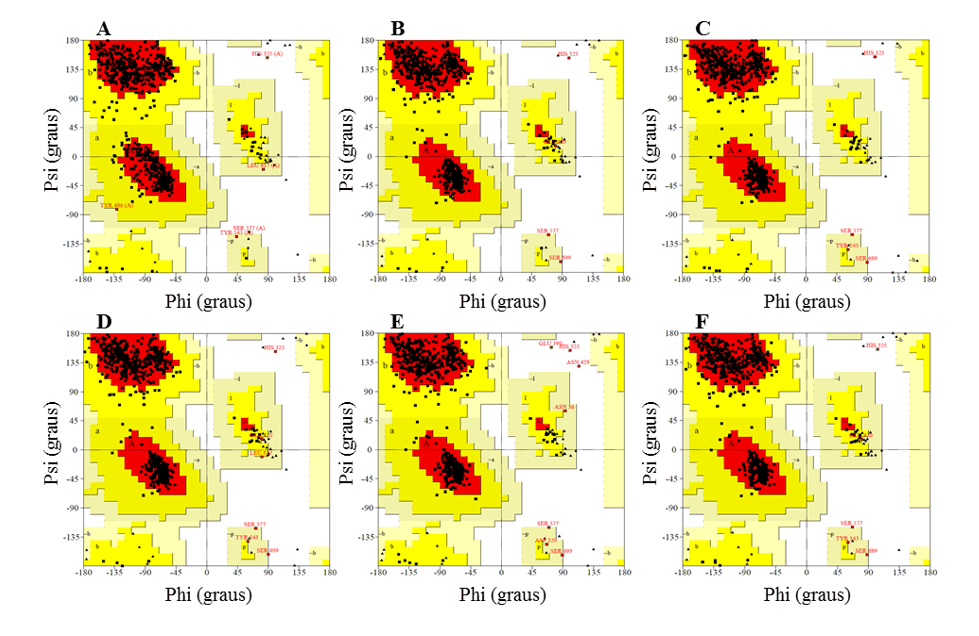
Au cours de l’étape de validation, ils ont été analysés sur la base d’analyses de graphiques ramachandran (fournies par le serveur PDBsum), de score 3D-1D (fourni par le programme verify-3D) et de score-Z (fourni par le serveur ProSA-web). Les valeurs obtenues pour les modèles ont été comparées à celles obtenues pour le moule.
Dans le graphique de Ramachandran, les modèles présentaient la majorité des résidus dans les régions favorables, allant de 91,2 à 92,3 %, tandis que le pourcentage de résidus dans les régions défavorables était d’un maximum de 0,5 %, et les meilleurs modèles étaient L.infantum et L.brasilienses. Ces modèles présentaient le plus grand nombre de résidus dans les régions favorables avec 92,3 % et 92,2 % et le plus faible pourcentage de résidus dans les régions défavorables avec respectivement 0,3 % et 0,2 % (figure 6).
Dans tous les modèles, il a été possible d’observer que le résidu de Ser, qui fait partie de la triade catalytique, se trouvait dans les régions défavorables. Cependant, ce fait n’affecte pas la validité des modèles, puisque lors de la comparaison du moule présenté le même résultat. Par conséquent, ce résultat ne configure pas une faible fiabilité des modèles.
Figure 6 : Résultats des graphiques ramachandran, obtenus par le programme PROCHECK, les structures des modèles de BPO générés et le moule.
| Structures | % de déchets dans les régions | ||||
| Favorable | Autorisé | Défavorable | |||
| OLIGOPEPTIDASE B | L. major (A)
(PDB 2XE4) |
90,2 | 9,5 | 0,3 | |
| L.brasilienses (B) | 92,2 | 7,7 | 0,2 | ||
| L. donovani (C) | 91,9 | 8,0 | 0,2 | ||
| L. infantum (D) | 92,3 | 7,3 | 0,3 | ||
| L. Mexicana (E) | 91,2 | 8,3 | 0,5 | ||
| L. panamensis (F) | 91,4 | 8,3 | 0,3 | ||
Source: Préparé par l’auteur sur la base des résultats procheck.
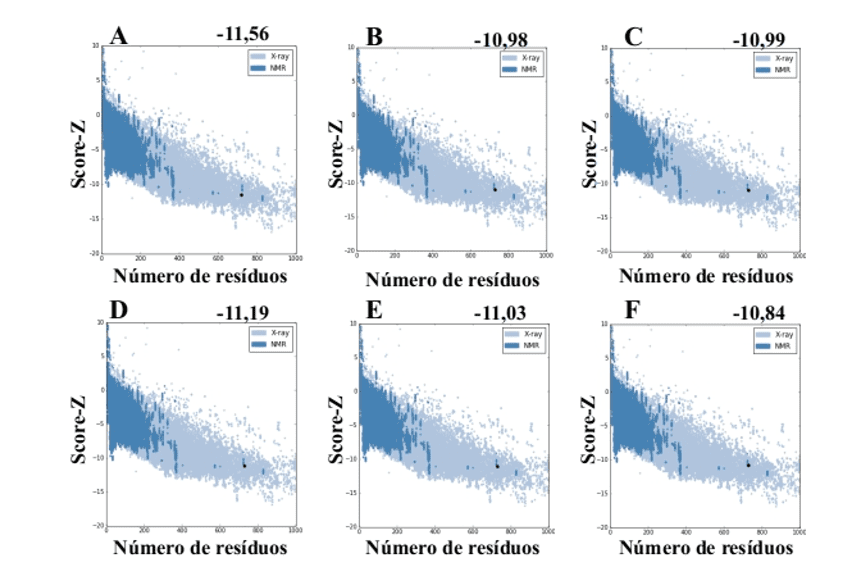
Lors de l’utilisation du serveur ProSA-web à partir des modèles générés par MODELLER, ils ont présenté des valeurs de score-Z -10.84 à -11.19 et ces valeurs sont compatibles avec les structures PDB (figure 7).
Figure 7 : Résultats de score Z calculés sur le serveur ProSA-web des structures de moule (à titre de comparaison). (A) L .major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. La région en bleu foncé indique la vingtaine des protéines obtenues par NMR et en bleu clair des protéines obtenues par diffraction aux rayons X.
Le programme Vérifier la 3D a été utilisé pour évaluer la compatibilité entre les structures 1D et 3D. Les modèles obtenus présentaient 95,28% à 98,34% des acides aminés avec une compatibilité 0,2 3D-1D, et les modèles L. donovani et L.brasilienses ont obtenu de meilleurs résultats. Selon les paramètres idéaux du programme, la plupart des résidus devraient présenter des valeurs supérieures à zéro, puisque les valeurs inférieures à zéro indiquent les régions de la molécule avec des problèmes. Le pourcentage d’acides aminés avec compatibilité 3D-1DMD 0,2 doit être supérieur à 80 % (EISENBERG et coll., 1997). Ainsi, ces résultats indiquent que les modèles ont présenté la compatibilité 1D-3D, et les résidus qui ont présenté l’incompatibilité ne font pas partie du site actif des enzymes (tableau 2).
Tableau 2 : Résultats de Vérifier 3D, montrant le pourcentage de résidus avec score de 0,2.
| Estruturas | % de resíduos com score > 0,2 | |
| OLIGOPEPTIDASE B | L.major
(PDB 2XE4) |
93,20 |
| L.brasilienses | 95,62 | |
| L.donovani | 97,12 | |
| L.infantum | 94,93 | |
| L.mexicana | 95,62 | |
| L.panamensis | 94,93 |
Source: Auteur.
VALIDATION DE LA TRIADE CATALYTIQUE DE L’ENZYME DES MODÈLES
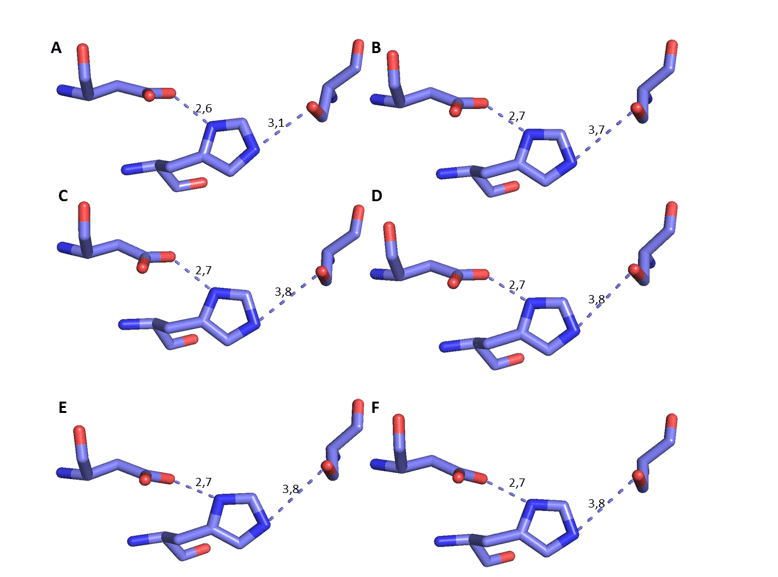
En raison du mécanisme de PB, il est essentiel d’analyser la distance et l’orientation entre les résidus d’acide aminé et la triade catalytique (Ser, Asp et His) des modèles générés afin d’augmenter la fiabilité des modèles.
Il est possible d’observer la comparaison de ces résidus spécifiques des modèles OPB avec le moule en question à la figure 17. En outre, il a été possible de mesurer la distance entre les résidus de la triade catalytique, en fonction du mécanisme d’interaction entre le site et le substrat, avec Son comme référence (DEREWENDA et al. , 1994). Par conséquent, il doit y avoir une distance spécifique au mécanisme d’action. Cette distance devrait être d’environ 3,5 euros entre son oxygène et l’azote Ser, en plus de la distance d’environ 2,6 euros entre l’oxygène aspartate et son autre azote, tel que décrit dans la littérature (DEREWENDA et al. , 1994). Dans cette analyse, il a été possible de vérifier que la distance en question a subi une petite variation, et est toujours conforme à ce qui a déjà été décrit. Cette distance est importante parce que les protéases sérinoes exigent que son clivage de substrat se produise (HEDSTROM, 2002) (figure 8). Sur la base de ces résultats fiables, la caractérisation des modèles a été poursuivie.
Figure 8 : Représentation 3D de la triade catalytique des BPO, respectivement, Ser, His et Asp. La couleur bleu foncé représente l’atome d’azote, l’oxygène rouge et le carbone lilas. Para OPB, (A) L.major (molde)e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis.
CARACTÉRISATION DES OLIGOPEPTIDASES DES LEISHMANIES
STRUCTURE SECONDAIRE DE PRÉDICTION
De la prédiction de la structure secondaire des enzymes des BPO, le nombre et la position des structures secondaires des modèles ont été révélés. Le PSISPRED a présenté entre 15 et 16 structures à hélice et tous présenté 38 paires de feuilles (tableau 3). D’après les résultats générés par PSIPRED, une comparaison visuelle de cette structure secondaire prévue avec les structures tridimensionnelles des 5 modèles a été effectuée, à l’aide du programme Pymol. Les modèles de L.brasilienses, et L. panamensis ont présenté la meilleure similitude, par rapport aux structures prédites secondaires et tertiaires (2D-3D), présentant 15 ‘-hélices et 38 ‘feuilles. Sur les 15 hélices présentées dans la structure tridimensionnelle du modèle 8 des hélices étaient dans la même position que prévu par le PSIPRED. Ces hélices correspondent à la séquence d’acides aminés : 59 à 74, 78 à 93, 535 à 540, 545 à 563, 631 à 640, 703 à 721 et 727 à 730.
Dans l’analyse générale, il n’y avait pas de différence exacerbée dans les quantités de feuilles d’hélice et de feuilles entre les prédictions et les modèles obtenus (tableau 3).
Tableau 3 : Comparaison entre les structures secondaires prédites par psipred et celle trouvée à travers Pymol.
| Modelos | α-Hélices | Folhas β | PSIPRED
α-Hélices |
PSIPRED
Folhas β |
| L.brasiliensis | 15 | 38 | 11 | 36 |
| L. donovani | 16 | 38 | 11 | 36 |
| L. infantum | 16 | 38 | 11 | 36 |
| L. mexicana | 15 | 38 | 10 | 36 |
| L. panamensis | 15 | 38 | 11 | 36 |
Source: Auteur.
De plus, il a été possible d’observer que le montant de 15 ‘-Propellers était le même pour L.brasilienses, L. Mexicana et L. panamensis. Tout comme L.infantum et L.donovani, ils ont obtenu le numéro 16 -Helix. Selon l’arbre phylogénétique, ces deux groupes mentionnés sont dans le même nœud interne et sont considérés comme monophylétiques (figure 2).
Ensuite, le RMSD (écart racine-moyen-carré) a été effectué entre les modèles et le moule. Les valeurs étaient prometteuses, comme on peut le voir dans le tableau 4, parce que les RMSD ne dépassaient pas la valeur de 0,19. Cette conclusion peut être justifiée par le degré élevé d’identité entre le moule et les modèles respectifs. En général, on s’attend à ce que les protéines d’identité supérieure à 30 %, aient un excellent chevauchement des principales chaînes, obtenant ainsi un RMSD de l’ordre de 2 ‘ (BENNER et al., 1997 ; CHOTHIA et coll., 1986).
Tableau 4 : RMSD des BPO générés par le Modeller, ayant avec orientation les carbones alpha du moule OPB de L. major.
| Molde | Modelos | RMSD (Å) |
| L. major
(2XE4) |
L.brasiliensis | 0,15 |
| L. donovani | 0,15 | |
| L. infantum | 0,16 | |
| L. Mexicana | 0,19 | |
| L. panamensis | 0,14 |
Source: Auteur.
CARTE DU POTENTIEL ÉLECTROSTATIQUE MOLÉCULAIRE (DÉPUTÉ) DE LA SURFACE DES ENZYMES ET DES SITES RÉCEPTIFS
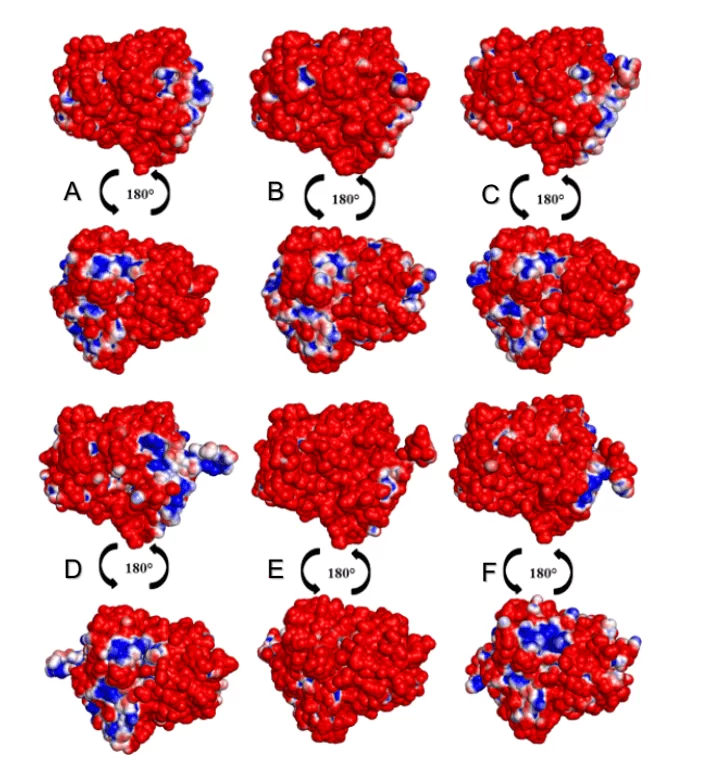
Dans l’analyse des eurodéputés sur les surfaces des BPO, il a été possible de constater que toutes les espèces de leishmania présentaient un pourcentage plus élevé de régions négatives que positives, comme le montre la figure 15. L’espèce L. donovani et L. infantum (groupe vert) a suggéré une zone négative (en couleur bleue) dans la même région. Les espèces L.brasilienses et L. panamensis (groupe rouge) ont présenté une région négative similaire, dans un modèle colorimétrique similaire. Les deux résultats peuvent être justifiés par le fait que l’espèce comparée à l’autre appartient au même monophylétique (figure 2).
Figure 9 : Carte du potentiel électrostatique des modèles 3D des BPO de leishmania spp et de celui de leur moule. Para OPB, (A) L.major (molde) e modelos: (B) L. brasilienses (C) L. donovani, (D) L. infantum, (E) L. mexicana (F) L. panamensis. Dans la couleur bleue, il présente la région positive et dans la couleur rouge, la région négative.
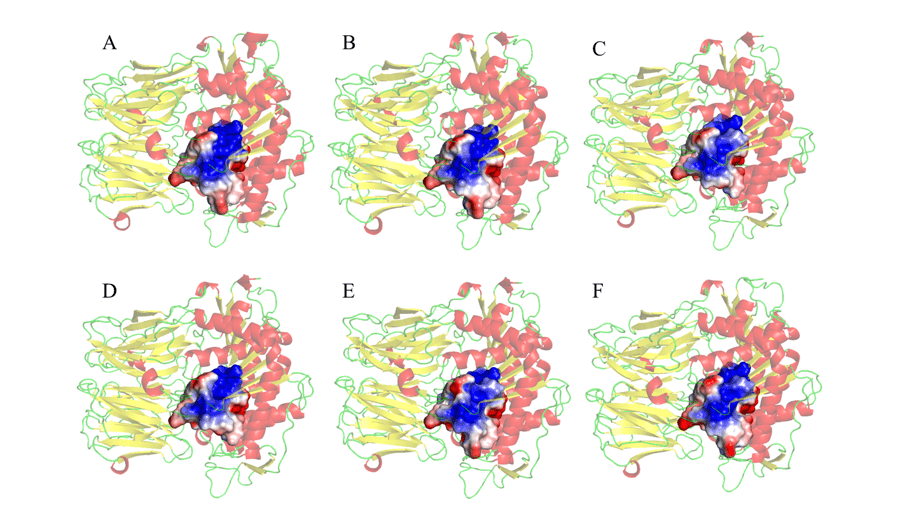
Enfin, le député européen des résidus a également été exécuté dans un rayon de 5 euros autour de la triade catalytique, comme le montre la figure 10. Le choix d’observer le député de la région autour de la triade catalytique a été parce qu’il s’agit de la deuxième région d’interaction entre l’enzyme et le substrat, dans lequel il y aura un hébergement (ANDERSSON et al., 2010). Ainsi, compte tenu de l’un des objectifs de l’étude, il a été possible de vérifier d’importantes similitudes et correspondances entre le moule et les modèles testés. Ces résultats, comme le montre la figure 10, ont révélé principalement une plus grande partie électropositive (en bleu) dans la région centrale des sites de connexion. Des régions négatives (en rouge) ont été observées dans les zones périphériques des eurodéputés étudiés. Ces résultats sont prometteurs, car il a été constaté que ces régions sont entièrement similaires dans tous les modèles, et peut aider au développement d’un médicament qui a la capacité d’agir spécifiquement dans tous les modèles étudiés.
Figure 10 : Représentation de la carte potentielle de la surface électrostatique des résidus d’acide aminé, 5 ‘ autour de la triade catalytique qui composent le site actif de l’enzyme. (A) L.major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. En rouge sont les hélices, vert les poignées et jaune les feuilles- . Dans la couleur dorée, le site de reliure. Dans la couleur bleue, il présente la région positive et dans la couleur rouge, la région négative.
DOGSITESCORER, CALIFORNIE
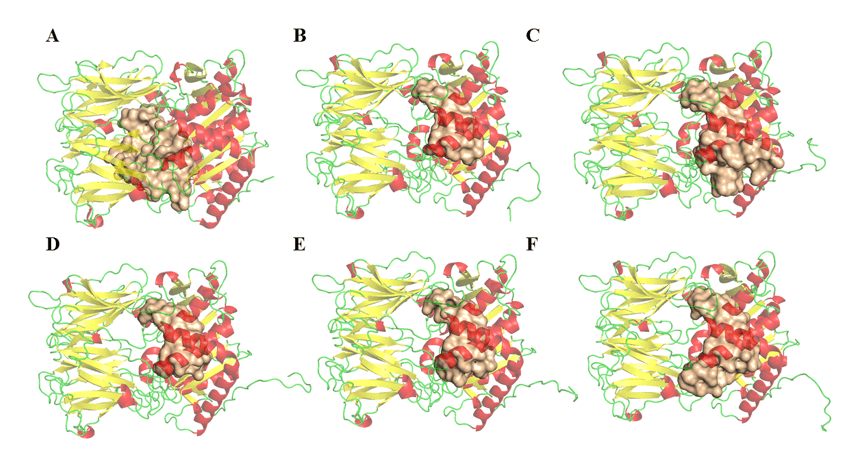
La détermination des paramètres de volume, de superficie et de profondeur des sites contraignants possibles des enzymes opb des espèces de Leishmania a été effectuée dans le programme DoGSiteScorer (http://poseview.zbh.uni-hamburg.de/) de ProteinsPlus – Structure-Based Modeling Support Server (VOLKAMER et al., 2012). Le programme souligne trois sites de liaison tels que décrits dans la littérature. La figure 11 montre la comparaison entre la région possible du site actif et ses différences. Ces variables parmi les BPO peuvent être liées à des résidus qui ne sont pas décrits comme importants pour leur inhibition, mais qui ont été pris en considération au cours de cette analyse. Ainsi, ce résultat n’exclut pas la possibilité que l’antipaine ou toute autre molécule ait un large spectre d’inhibition sur l’enzyme, en raison du fait que les résultats liés à drugscore étaient très similaires et positifs dans toutes les régions. Le tableau 5 est observé, un écart relatif entre le volume et les résultats superficieaux des cavités trouvées dans les modèles. Ce résultat ne suggère pas un biais de doute sur le potentiel inhibiteur d’un seul médicament dans les enzymes respectives. Cependant, il prête attention au fait que certaines protéines ont des cavités plus grandes que d’autres. Cependant, ces mêmes cavités ont des points communs, et ce sont les résidus qui composent le site catalytique, comme on peut le voir dans la figure 11 et peuvent être explorés.
Tableau 5 : Valeurs se référant aux régions de connexion possibles des BPO (obtenues par le serveur).
| Estruturas | DogSiteScoore | |||
| Volume | Área | Drug Score | ||
| OLIGOPEPTIDASE B | L. major
(PDB 2XE4) |
1690,62 | 1818,41 | 0,80 |
| L.brasiliensis | 1527,84 | 1766,63 | 0,80 | |
| L. donovani | 1074,57 | 1428,55 | 0,79 | |
| L. infantum | 1309,97 | 1572,19 | 0,80 | |
| L. mexicana | 800,92 | 799,38 | 0,85 | |
| L. panamensis | 971,96 | 1083,40 | 0,81 | |
Source: Auteur.
Figure 11 : Structures de BPO et régions de connexion possibles (obtenues par le serveur DogSite). (A) L .major (molde)e modelos: (B) L. brasilienses, (C), L. donovani, (D) L. infantum, (E) L. mexicana e (F) L. panamensis. En rouge sont les hélices, vert les poignées et jaune les feuilles- . Dans la couleur dorée, le site de reliure.
MODES NORMAUX
Après la caractérisation de l’enzyme dans les sites structurels, superficiels et contraignants, les modes normaux pour les enzymes de chaque espèce ont été exécutés dans le but de leurs mouvements respectifs.
Après la relaxation et l’énergie minimizaçàes effectuées par la dynamique moléculaire dans GROMACS, ces structures ont été soumises à des analyses normales afin d’observer d’éventuels mouvements compatibles.
Il a été possible d’observer dans tous les modèles étudié un mouvement expressif d’une hélice spécifique, et lors de l’analyse de cette région dans sa composition en acides aminés, il est possible de remarquer qu’il est fortement conservé. Ainsi, le modèle de mouvement a été répété dans tous les modèles en question. Cette région suggéra un mouvement linéaire s’éloignant du centre vers la périphérie, exposant la triade catalytique. Cela peut être révélateur du mouvement effectué par la protéine pour l’hébergement du substrat.
Lors de l’observation de la figure 12 et 13, le même motif de couleur peut être observé dans tous les modèles de l’étude. Ces données représentent la capacité donnée à la région d’aller dans une certaine direction. Les couleurs bleues représentent des régions plus invariables au cours de la simulation par des modes normaux, avec cela, un modèle de rigidité dans les protéines est perceptible correspondant principalement aux feuilles du domaine de l’hélice et quelques hélices qui composent le domaine catalytique. Les couleurs proches du vert représentent des régions intermédiaires par rapport à la capacité et à l’amplitude de mouvement. Sur la base de ces informations, il est possible de percevoir cette coloration présente en boucles aux extrémités des modèles ainsi que dans certaines hélices du domaine catalytique. Enfin, il y a ces régions qui ont présenté une coloration orange/rouge, dans laquelle est la représentation d’un énorme potentiel de mouvement. Ainsi, on peut voir qu’il y a de petites régions de boucles aux extrémités des domaines catalytiques avec ce potentiel de mouvement, ainsi qu’une hélice située dans la partie du domaine catalytique, plus spécifiquement devant la triade catalytique des BPO, dans laquelle elle a obtenu le mouvement le plus expressif de l’étude.
Tous les modèles ont obtenu la même configuration de mouvement, où une hélice (en orange) s’est avérée être la région avec la plus grande capacité de mouvement. Ainsi, les BPO de L. brasiliensis, L. donovani, L. infantum, L. mexicana et L. panamensis dans leur plus grande gamme de mouvements obtenus, respectivement, 9,8 , 11,3 , 11,8 , 11,9 et 11,6 ‘(figure 12 et 13).
Figure 12 : Résultat de l’analyse par des modes normaux de représentation par le programme de pymol, dans l’image les mouvements des modèles sont démontrés, dans leurs formes détendues (droite) par rapport au mouvement d’une plus grande amplitude (à gauche). Sont également représentées les régions les plus rigides (bleu foncé), les régions avec peu de mouvement (bleu clair), les régions intermédiaires (vert), les régions à fort potentiel de mouvement (Orange) et les régions extrêmement malléables (rouge) (Partie1/2).
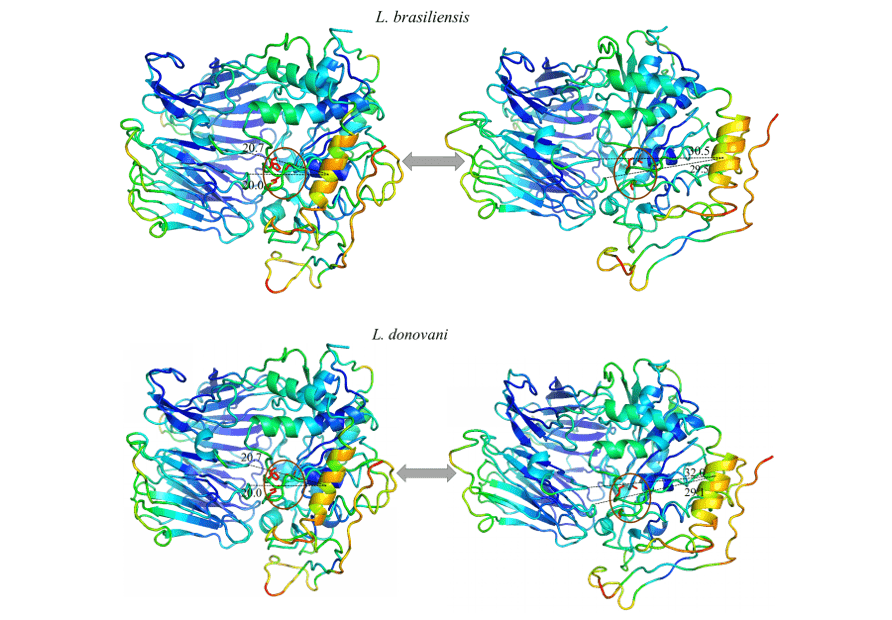
Figure 13 : Résultat de l’analyse par des modes normaux de représentation par le programme de pymol, dans l’image les mouvements des modèles sont démontrés, dans leurs formes détendues (droite) par rapport au mouvement d’une plus grande amplitude (à gauche). Sont également représentées les régions les plus rigides (bleu foncé), les régions avec peu de mouvement (bleu clair), les régions intermédiaires (vert), les régions à fort potentiel de mouvement (Orange) et les régions extrêmement malléables (rouge). (Partie 2/2)
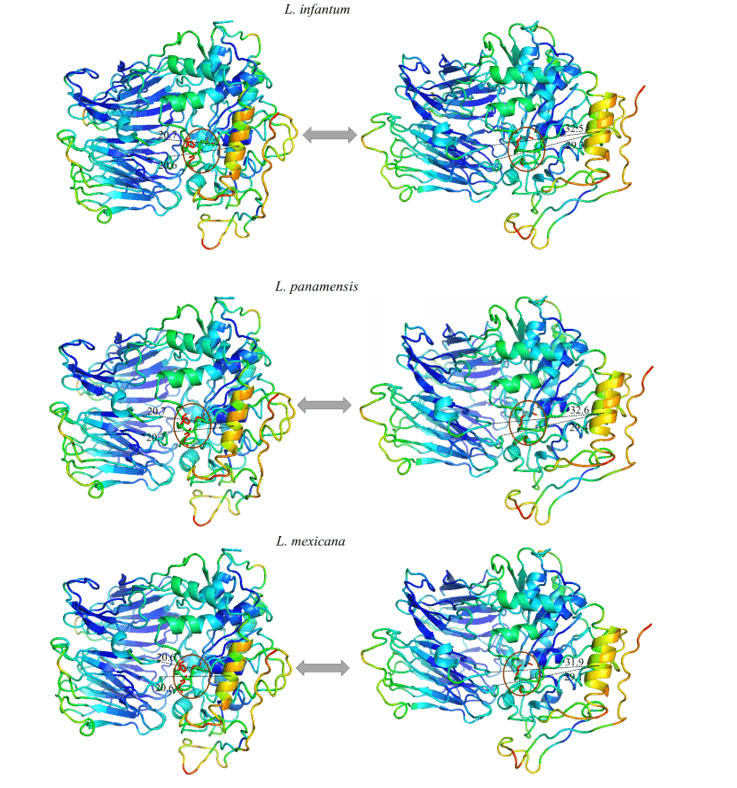
Dans ce cas, les études normales approuvent un mécanisme possible de BPO qui n’est pas encore décrit pour les espèces de leishmania spp. jusqu’à la présente étude.
CONCLUSION
Dans cette étude, les modèles tridimensionnels de l’enzyme opb de L.brasiliensis, L. donovani, L. infantum, L. mexicana et L. panamensis ont été obtenus. La validation des modèles a présenté des résultats fiables pour tous les modèles tridimensionnels obtenus. Dans la caractérisation de l’enzyme, la carte potentielle électrostatique de surface a montré que la plupart des résidus présentaient une charge négative. Dans la caractérisation de la région autour de la triade catalytique, elle a démontré la similitude entre le volume, la zone et la correspondance entre les résidus positifs et négatifs. Par conséquent, il a été possible de vérifier que les résultats de l’analyse par des modes normaux suggéraient un mouvement expressif dans l’une des hélices spécifiques, se produisant une distance linéaire de ceci, du centre vers la périphérie, exposant ainsi la triade catalytique. La description de ces mouvements effectués par cette enzyme est d’une grande importance pour aider à comprendre son fonctionnement.
Enfin, les résultats de la présente étude peuvent ajouter des connaissances à la communauté scientifique, apportant des élucidations et de nouvelles questions liées au thème, servant de base pour d’éventuelles études dans le domaine de la santé.
RÉFÉRENCES
A. Benner S, Cannarozzi G, Gerloff D, Turcotte M, Chelvanayagam G. Bona Fide Predictions of Protein Secondary Structure Using Transparent Analyses of Multiple Sequence Alignments. Chem Rev. 1997;97(8):2725-2844. doi:10.1021/cr940469a.
Alva, V., Nam, S. Z., Söding, J., & Lupas, A. N. (2016). The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Research, 44(W1), W410–W415. doi.org/10.1093/nar/gkw348.
Alvarenga DG, Escalda PMF, da Costa ASV, Monreal MTFD. Leishmaniose visceral: Estudo retrospectivo de fatores associados à letalidade. Rev Soc Bras Med Trop. 2010;43(2):194-197.
Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389‐3402. doi:10.1093/nar/25.17.3389.
Andersson CD, Chen BY, Linusson A. Mapping of ligand-binding cavities in proteins [published correction appears in Proteins. 2011 Apr;79(4):1363]. Proteins. 2010;78(6):1408–1422. doi:10.1002/prot.22655.
Bailey F, Mondragon-Shem K, Hotez P, et al. A new perspective on cutaneous leishmaniasis-Implications for global prevalence and burden of disease estimates. PLoS Negl Trop Dis. 2017;11(8):e0005739. Published 2017 Aug 10. doi:10.1371/journal.pntd.0005739.
Carmo RF, Luz ZMP da, Bevilacqua PD. Percepções da população e de profissionais de saúde sobre a leishmaniose visceral. Cien Saude Colet. 2016;21(2):621-628. doi:10.1590/1413-81232015212.10422015.
Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5(4):823‐826.
Derewenda ZS, Derewenda U, Kobos PM. (His)Cε-H···O=C< Hydrogen Bond in the Active Sites of Serine Hydrolases. J Mol Biol. 1994;241(1):83-93. doi:10.1006/JMBI.1994.1475.
Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32(Web Server issue):W665–W667. doi:10.1093/nar/gkh381.
Eisenberg, D., Lüthy, R., & Bowie, J. U. (1997). [20]VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods in Enzymology, 277, 396–404. https://doi.org/10.1016/S0076-6879(97)77022-8.
Eyal E, Lum G, Bahar I. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics. 2015;31(9):1487–1489. doi:10.1093/bioinformatics/btu847.
Ghorbani Masoud, Farhoudi Ramin. Leishmaniasis in humans: drug or vaccine therapy? Drug Des Devel Ther. 2018;12:25-40. doi:10.2147/DDDT.S146521.
Hedstrom L. Serine Protease Mechanism and Specificity. Chem Rev. 2002;102(12):4501-4524. doi:10.1021/cr000033x.
Katsila T, Spyroulias GA, Patrinos GP, Matsoukas MT. Computational approaches in target identification and drug discovery. Comput Struct Biotechnol J. 2016;14:177–184. Published 2016 May 7. doi:10.1016/j.csbj.2016.04.004.
Kumar S, Tamura K, Nei M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5(2):150-163.
Macedo-Silva RM, dos Santos C de LP, Diniz VA, De Carvalho JJ, Guerra C, Côrte-Real S. Peripheral blood fibrocytes: New information to explain the dynamics of Leishmania infection. Mem Inst Oswaldo Cruz. 2014;109(1):61-69. doi:10.1590/0074-0276130247
Machado P de A, Carneiro MPD, Sousa-Batista A de J, et al. Leishmanicidal therapy targeted to parasite proteases. Life Sci. 2019;219:163-181. doi:10.1016/J.LFS.2019.01.015.
Morris, A. L., MacArthur, M. W., Hutchinson, E. G., & Thornton, J. M. (1992). Stereochemical quality of protein structure coordinates. Proteins: Structure, Function, and Bioinformatics, 12(4), 345–364. https://doi.org/10.1002/prot.340120407.
Ovchinnikova M V., Mikhailova AG, Karlinsky DM, Gorlenko VA, Rumsh LD. Reversible cyclic thermal inactivation of oligopeptidase B from Serratia proteamaculans. Acta Naturae. 2018;10(2):65-70.
Ramachandran, G. N., Ramakrishnan, C., & Sasisekharan, V. (1963). Stereochemistry of polypeptide chain configurations. Journal of Molecular Biology, 7(1), 95–99. https://doi.org/10.1016/S0022-2836(63)80023-6.
Santos Filho, O. A., & Alencastro, R. B. de. (2003). Modelagem de proteínas por homologia. Química Nova, 26(2), 253–259. https://doi.org/10.1590/S0100-40422003000200019
Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. Published 2011 Oct 11. doi:10.1038/msb.2011.75.
SODERO ACR, DOS SANTOS ACGO, MELLO JFRE, et al. Oligopeptidase B and B2: comparative modelling and virtual screening as searching tools for new antileishmanial compounds. Parasitology. 2017;144(4):536-545. doi:10.1017/s0031182016002237.
Swenerton RK, Zhang S, Sajid M, et al. The oligopeptidase B of Leishmania regulates parasite enolase and immune evasion. J Biol Chem. 2011;286(1):429-440. doi:10.1074/jbc.M110.138313.
Volkamer, A., Kuhn, D., Rippmann, F., & Rarey, M. (2012). Dogsitescorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics, 28(15), 2074–2075. https://doi.org/10.1093/bioinformatics/bts310.
Wang Q, Arighi CN, King BL, et al. Community annotation and bioinformatics workforce development in concert–Little Skate Genome Annotation Workshops and Jamborees. Database (Oxford). 2012;2012:bar064. Published 2012 Mar 20. doi:10.1093/database/bar064
Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics. 2016;54:5.6.1–5.6.37. Published 2016 Jun 20. doi:10.1002/cpbi.3.
Wiederstein, M., & Sippl, M. J. (2007). ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research, 35(SUPPL.2), 407–410. https://doi.org/10.1093/nar/gkm290.
WHO. Integrating Neglected Tropical Diseases into Global Health and Development: Fourth WHO Report on Neglected Tropical Diseases.; 2017. http://apps.who.int/iris/bitstream/10665/255011/1/9789241565448-eng.pdf?ua=1.
WHO. (2019). Leishmanioses – Informe Epidemiológico das Américas No 7 – Março, 2019. Retrieved from http://iris.paho.org/xmlui/bitstream/handle/123456789/50505/ 2019-cde-leish-informe-epi-das-americas.pdf?sequence=2&isAllowed=y.
ANNEXE – CHIFFRES ET TABLEAUX EN ANGLAIS
Schéma 1 : Schéma simplifié des étapes et des méthodes matérielles.
Tableau 1 : Pourcentage d’identité entre les modèles d’oligopeptité B des espèces de Leishmania et leur moule respectif.
| Mold protein (code PDB) | Models OPB
(código uniprot) |
Identity (%) | Gaps (%) |
| OPB
L. major (code PDB 2XE4) |
L.brasiliensis
(A4H5Q8) |
86 | 0 |
| L. donovani
(C9EF60) |
96 | 0 | |
| L. infantum
(A4HTZ8) |
96 | 0 | |
| L. Mexicana
(E9AMS8) |
90 | 0 | |
| L. panamensis
(A0A088RJA7) |
86 | 0 |
Fonte: Autoral.
Figure 6 : Résultats des graphiques ramachandran, obtenus par le programme PROCHECK, les structures des modèles de BPO générés et le moule.
| Structures | % waste in the regions | ||||
| Favorable | Allowed | Unfavorable | |||
| OLIGOPEPTIDASE B | L. major (A)
(PDB 2XE4) |
90,2 | 9,5 | 0,3 | |
| L.brasilienses (B) | 92,2 | 7,7 | 0,2 | ||
| L. donovani (C) | 91,9 | 8,0 | 0,2 | ||
| L. infantum (D) | 92,3 | 7,3 | 0,3 | ||
| L. Mexicana (E) | 91,2 | 8,3 | 0,5 | ||
| L. panamensis (F) | 91,4 | 8,3 | 0,3 | ||
Source: Préparé par l’auteur sur la base des résultats procheck.
Figure 7 : Résultats de score Z calculés sur le serveur ProSA-web des structures de moule (à titre de comparaison). (A) L .major (molde) e modelos: (B) L. brasilienses (C) L. donovani (D) L. infantum (E) L. mexicana (F) L. panamensis. La région en bleu foncé indique la vingtaine des protéines obtenues par NMR et en bleu clair des protéines obtenues par diffraction aux rayons X
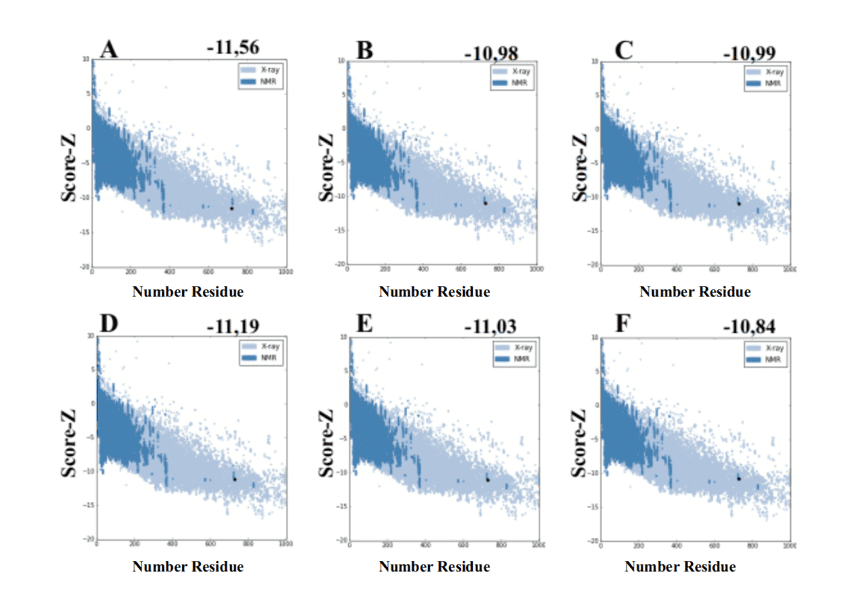
Tableau 2 : Résultats de Vérifier 3D, montrant le pourcentage de résidus avec score de 0,2.
| Structures | % residue with score > 0,2 | |
| OLIGOPEPTIDASE B | L.major
(PDB 2XE4) |
93,20 |
| L.brasilienses | 95,62 | |
| L.donovani | 97,12 | |
| L.infantum | 94,93 | |
| L.mexicana | 95,62 | |
| L.panamensis | 94,93 |
Source: Auteur.
Tableau 3 : Comparaison entre les structures secondaires prédites par psipred et celle trouvée à travers Pymol.
| Models | α-Hélix | Sheet β | PSIPRED
α-Hélix |
PSIPRED
Sheet β |
| L.brasiliensis | 15 | 38 | 11 | 36 |
| L. donovani | 16 | 38 | 11 | 36 |
| L. infantum | 16 | 38 | 11 | 36 |
| L. mexicana | 15 | 38 | 10 | 36 |
| L. panamensis | 15 | 38 | 11 | 36 |
Source: Auteur.
Tableau 4 : RMSD des BPO générés par le Modeller, ayant avec orientation les carbones alpha du moule OPB de L. major.
| Mold | Models | RMSD (Å) |
| L. major
(2XE4) |
L.brasiliensis | 0,15 |
| L. donovani | 0,15 | |
| L. infantum | 0,16 | |
| L. mexicana | 0,19 | |
| L. panamensis | 0,14 |
Source: Auteur.
Tableau 5 : Valeurs se référant aux régions de connexion possibles des BPO (obtenues par le serveur).
| Structures | DogSiteScoore | |||
| Volume | Area | Drug Score | ||
| OP OLIGOPEPTIDASE B | L. major
(PDB 2XE4) |
1690,62 | 1818,41 | 0,80 |
| L.brasiliensis | 1527,84 | 1766,63 | 0,80 | |
| L. donovani | 1074,57 | 1428,55 | 0,79 | |
| L. infantum | 1309,97 | 1572,19 | 0,80 | |
| L. mexicana | 800,92 | 799,38 | 0,85 | |
| L. panamensis | 971,96 | 1083,40 | 0,81 | |
Source: Auteur.
[1] Biomédicale et maîtrise en biochimie médicale.
[2] Master en sciences pharmaceutiques et pharmaceutiques de l’UFRJ.
[3] Doctorat en chimie, maîtrise en chimie organique et produits pharmaceutiques industriels.
Envoyé : Mai 2020.
Approuvé : mai 2020.