ARTIGO DE REVISÃO
BARBOSA, Jhuliene Carla [1], OLIVEIRA, Rodolfo Lee Carvalho de [2], LIMA, Yuri Correia de [3], JÚNIOR, Roberto Belo Cardoso [4], BARROS, Danilo Pontes de Oliveira [5]
BARBOSA, Jhuliene Carla. Et al. Tratamento medicamentoso de episódios hemorrágicos em procedimentos cirúrgicos nos pacientes portadores da Doença de Von Willebrand. Revista Científica Multidisciplinar Núcleo do Conhecimento. Ano 05, Ed. 09, Vol. 05, pp. 136-154. Setembro de 2020. ISSN: 2448-0959, Link de acesso: https://www.nucleodoconhecimento.com.br/saude/episodios-hemorragicos
RESUMO
A doença de Von Willebrand (DvW), se apresenta como um sangramento na mucosa devido a qualidade ou quantidade diminuída do fator de Von Willebrand (FvW), uma proteína adesiva plaquetária. O diagnóstico da DvW necessita de uma avaliação mais cautelosa de fatores individuais do paciente, episódios de sangramento e resultados laboratoriais. O objetivo foi discorrer sobre as opções terapêuticas medicamentosas relacionadas aos pacientes portadores da doença no período perioperatório. Tratou-se de uma revisão de literatura bibliográfica com estratégia qualitativa, de caráter narrativo, realizada a partir da coleta de artigos publicados em bases de dados como os sites da Scientific Eletronic Library Online (Scielo), Literatura Internacional em Ciências da Saúde (PubMed/MEDLINE) e no banco de dados da Literatura Latino-Americana e do Caribe em Ciências da Saúde (LILACS). A pesquisa foi realizada a partir das palavras-chaves incluídas no Descritores em Ciências da Saúde (DeCS), relacionadas ao assunto principal e ao foco requerido no estudo: “Doença von Willebrand”, “Fator von Willebrand”, “Procedimentos cirúrgicos” e “Terapia medicamentosa”. Foram considerados artigos publicados no período de 2008 a 2020, disponíveis em língua portuguesa, inglesa e espanhola. O tratamento do sangramento na DvW envolve o uso de desmopressina e concentrados de FvW derivados do plasma e uma variedade de agentes adjuvantes. Finalmente, a compreensão sobre a patologia visando aplicação eficaz das terapêuticas disponíveis são, portanto, essenciais para pacientes submetidos a grandes cirurgias, particularmente com formas mais graves de DvW.
Palavras-chave: Doença von Willebrand, Fator von Willebrand, procedimentos cirúrgicos, terapia medicamentosa.
INTRODUÇÃO
A doença de von Willebrand (DvW) é um distúrbio hemorrágico hereditário de traço autossômico dominante, provocado por uma modificação quantitativa ou qualitativa do fator von Willebrand (FvW), uma glicoproteína plasmática adesiva multimérica grande, que exerce uma função fundamental na hemostasia primária e secundária. (PINHEIRO et al., 2017), (CASTAMAN, 2017).
Está entre as desordens da coagulação hereditária mais frequente em humanos, afetando em média 1% da população mundial. (CAVALCANTI et al., 2018). A baixa precisão na avaliação da prevalência da doença é relacionada a complexidade do diagnóstico, devido a sintomatologia versátil e inespecífica e pela dificuldade de interpretação nos exames laboratoriais. (SANTOS, 2017).
Essa patologia tem três classificações primárias: tipo 1, 2 e 3. O tipo 1 corresponde a deficiência quantitativa parcial de FvW, o tipo 2 a deficiência qualitativa, este é subdividido em quatro subtipos 2A, 2B, 2M e 2N, conforme o sitio funcional em que a anormalidade está localizada. (ECHAHDI et al., 2017). O tipo 3, forma mais severa, é relacionado a deficiência praticamente completa. A expressão clínica da DvW é de modo geral leve no tipo 1, com gravidade progressiva no tipo 2 e tipo 3. (HEIJDRA; CNOSSEN, 2017).
Os pacientes geralmente são diagnosticados a partir de sangramentos cutâneos e mucosos, histórico familiar de ocorrências hemorrágicas e irregularidades laboratoriais no FvW, factor VIII de coagulação (FVIII) ou ambos. Os testes regulares para triagem da consistem na verificação dos níveis de FvW existentes no plasma e na eficiência desse fator em unir plaquetas quando o antibiótico ristocetina estiver presente. (NG et al., 2015). A junção desses testes, mais a avaliação do FVIII, possibilita a classificação dos portadores nos grupos tradicionais. São utilizados ainda alguns testes mais específicos para identificar os subtipos da doença, que indicará alternativas terapêuticas quando necessário, (LAFFAN et al., 2014).
Não é raro diagnostico após trauma ou cirurgia, a presença de hemorragia mais constante é o sangramento após uma extração dentária, sangramento pós-traumático excessivo e podem ocorrer sangramento pós-parto. Esses episódios e suas intensidades são normalmente equivalentes ao nível de deficiência dos dois fatores em questão. (SWAMI; KAUR, 2017).
O tratamento na DvW abrange o uso terapias não concentradas como pró-coagulantes, terapia concentrada e a terapêutica de transfusão com produtos derivados de plasma FvW e FVIII da coagulação. (BRASIL, 2018). Normalmente, o propósito do tratamento em pacientes é reparar as falhas na hemóstase, em ocorrências de aparições hemorrágicas e antes e durante procedimentos cirúrgicos. A prevenção e o tratamento podem reduzir os prejuízos físicos e melhorar o bem estar do paciente. (CASTAMAN et al., 2013).
O grau de aparições hemorrágicas e sua magnitude podem variar entre os indivíduos, a forma mais adequada de tratamento depende exclusivamente da classificação desse distúrbio. A diversidade de subtipos evidencia um obstáculo na escolha do melhor tratamento de forma única para cada paciente submetido a cirurgias. (LAVIN; O’DONNELL, 2016).
Levando em consideração o risco aumentado de episódios hemorrágicos prolongados ou excessivos, esses pacientes requerem um cuidado especial e um diagnóstico cuidadoso. Dessa maneira, conhecer as características da doença possibilita a classificação de pacientes com necessidade de uma terapia medicamentosa típica para prevenir ou tratar o sangramento, diminuindo as chances de complicações em procedimentos cirúrgicos.
Sendo assim, a pesquisa tem por objetivo discorrer sobre os aspectos em geral da doença e os medicamentos indicados aos pacientes portadores da DvW no período perioperatório.
MÉTODOS
Este estudo foi fundamentado em uma estratégia qualitativa de pesquisa, de caráter narrativo, por meio de revisão de literatura a partir da coleta de artigos publicados em bases de dados como os sites da Scientific Eletronic Library Online (Scielo), Literatura Internacional em Ciências da Saúde (PubMed/MEDLINE) e no banco de dados da Literatura Latino-Americana e do Caribe em Ciências da Saúde (LILACS). Os termos utilizados isoladamente ou em combinação para busca da bibliografia foram: “terapia medicamentosa”, “procedimentos cirúrgicos”, “doença von Willebrand”, “fator von Willebrand”. Foram utilizados 54 artigos publicados entre o ano de 2008 a 2020, nos idiomas português, inglês e espanhol. Os artigos foram lidos e considerados de acordo com a compatibilidade dos objetivos da pesquisa, desprezando aqueles que, independente de aparecerem nos resultados de busca, não expressaram o tema sob a perspectiva do estudo.
HEMOSTASIA: CONTROLE DO FVW
Os distúrbios hereditários da coagulação provocam deficiências na estrutura e na quantidade de uma ou mais proteínas plasmáticas. A hemostasia é um sistema de proteção do organismo, essencial para manter a integridade do vaso, evitar uma hemorragia em casos de lesão vascular e reparar a fluidez do sangue quando ela for restaurada. O seu funcionamento envolve quatro sistemas, que atuam de maneira especifica, ampliada e articulada, que são: sistema plaquetário, vascular, fibrinolítico e da coagulação. (GUERRERO; LÓPEZ, 2015).
O sistema da coagulação é formado por várias proteínas plasmáticas, como o exemplo do FvW, que desempenha uma atribuição fundamental. (VAN DIEVOET et al., 2020). O gene que define em numerosa parte os níveis de circulação desse fator, está localizado no cromossomo 12. (LEEBEEK et al., 2019). Nas células endoteliais, os corpos de Weibel-Palade são as organelas que o armazenam, ele é secretado nos vasos sanguíneos por exocitose, onde se reorganiza estruturalmente em encadeamento para reter plaquetas e dar início a hemostasia primária. (LENTING et al., 2015).
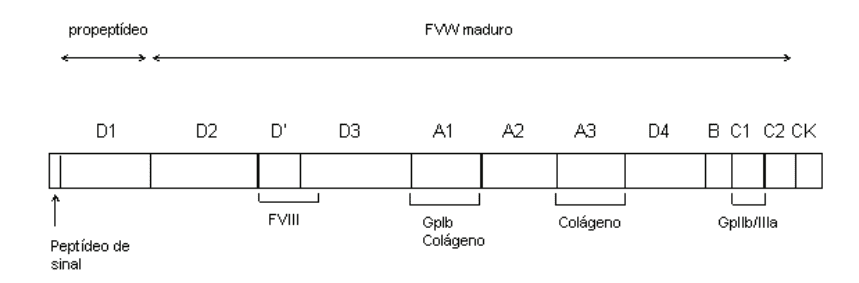
Os domínios D1 e D2 constituem esse propeptídeo e são quebrados na sua maturação. Os domínios D ‘e D3 se ligam ao FVIII, que ativado, pode então ativar o fator X, essa ativação da cascata de coagulação tem como resultado a produção de um coágulo de fibrina. (BERNTORP et al., 2018). O domínio A1 se une a plaquetas e colágeno, e o domínio A3 também se une ao colágeno. Assim, essa proteína desempenha um papel fisiológico essencial na adesão plaquetária. (KEESLER; FLOOD, 2017). A figura 1 representa a sua estrutura do.
Figura 1. Estrutura do Fator de Von Willebrand
Como constituinte da matriz extracelular (MEC), das células do endotélio, esse fator é capaz de desencadear de modo direto a agregação plaquetária. Quando livre, beneficia sua atuação na matriz, unindo-se as estruturas que ficam em contato com o fluxo de sangue, sobretudo o colágeno tipo I e III em níveis mais profundos do vaso, em companhia do colágeno microfibrilar tipo IV no subendotelio. Ademais, ele é fundamental para dar suporte a agregação plaquetas-plaquetas, promovendo aumento e solidificação do trombo. (RUGGERI; MENDOLICCHIO, 2015).
É uma alteração grandemente heterogênea, como afirmado pelo fato do FvW exercer várias utilidades adesivas que concedem a prisão e localização eficiente de plaquetas e FVIII nos vasos danificados, e exerce essa função porque consegue se unir a plaquetas através de inúmeros receptores como a glicoproteína Ib (GPIb), componentes da matriz, destacando-se o colágeno e também o FVIII que preserva sua função. Por essa razão as alterações podem ocorrer em seja qual for o lugar da estrutura desse fator, o que leva a uma ampla variedade de subtipos clínicos e laboratoriais da doença. (FAVALORO; SOMA, 2016).
CLASSIFICAÇÃO DA DVW E MÉTODOS DIAGNÓSTICOS
Pode ser diferenciada em seis tipos: 1, 2A, 2B, 2M, 2N e 3, de acordo com características fenotípicas genotípicas, clínicas e laboratoriais distintas. As manifestações clínicas mais comuns são: epistaxe, menorragia, sangramento gengival, hemorragia gastrintestinal, hemartrose e sangramento prolongado após procedimentos cirúrgicos. (RUGGERI; MENDOLICCHIO, 2015).
O tipo 1 da doença é causador de uma notável quantidade de casos: 70% a 80%. Há uma diversidade fenotípica considerável entre esses portadores, o que possivelmente é esperado, já que os níveis de antígenos FvW no plasma nesses pacientes oscilam entre 1 e 50 UI/ dl (faixa normal de 50-200 UI / dL). (LAVIN et al., 2017).
Os portadores do tipo 2 contribuem com aproximadamente 20% dos casos e procedem da expressão de uma molécula funcionalmente irregular. Por fim, pacientes raros do tipo 3 ± 0,5%, 1 por milhão de habitantes e resultam de casos onde os indivíduos podem ter praticamente a privação total. (LAVIN et al., 2017). As manifestações clínicas dos fenótipos da doença podem ser visualizadas no quadro 1.
Quadro 1. Manifestações clínicas da doença de von Willebrand
| Tipo da doença | Manifestações clínicas |
|
DvW tipo 1 |
Normalmente se apresenta como sangramento monocutâneo leve, porém pode ter agravantes quando os níveis de FvW são menores que 15 UI / dL. Sangramento ou hemorragia nasal são sintomas populares em crianças. Hipermenorreia é o evento mais manifesto em mulheres em idade fértil. (ECHAHDI et al., 2017). |
|
DvW tipo 2A |
Indivíduos com DVW tipo 2A geralmente apresentam sangramento monocutâneo leve a moderado. (HEIJDRA; CNOSSEN, 2017). |
|
DvW tipo 2B |
Os indivíduos geralmente apresentam sangramento monocutâneo leve a moderado. Uma característica que pode ocorrer é um agravamento da trombocitopenia durante situações estressantes, como infecção grave, durante cirurgia, gravidez, ou se tratado com desmopressina. (JAMES; GOODEVE, 2011). |
|
DvW tipo 2M |
Os indivíduos geralmente apresentam sintomas de sangramento monocutâneo leve a moderado, mas os episódios de sangramento podem ser graves, principalmente na presença de FvW muito baixo ou ausente. (DORUELO AL et al., 2017). |
|
DvW tipo 2N |
Os sintomas são essencialmente os mesmos que os observados na hemofilia leve A e incluem sangramento excessivo no momento da cirurgia ou procedimentos, pois ambos os distúrbios resultam da redução do FVIII. (GOODEVE, 2016). |
|
DvW tipo 3 |
Manifesta-se com sangramento grave, incluindo sangramento monocutâneo excessivo e sangramento musculoesquelético. (LAFFAN et al., 2014). |
Os tipos 1 e 3 são defeitos quantitativos, no primeiro caso, expressa a diminuição dos multímeros, todavia, sua função é mantida, as mutações são versáteis, refletidas por todo o gene, nesse exemplo, tanto o antígeno do fator de von Willebrand como a atividade coagulante do fator VIII estarão diminuídos de forma proporcional, acarretando uma deficiência leve ou moderada. (EL OUAALITI et al., 2016).
A deficiência que resulta no tipo 3 é uma grave carência, normalmente por conta da herança autossômica recessiva ou formada por um alelo nulo. Além de ocasionar as baixas taxas desse fator também contribui para baixos níveis do FVIII, que geralmente são <10 UI / dL, os padrões de sangramentos são graves e podem se assemelhar a hemofilia. (NG et al., 2015).
No tipo 2 o defeito é qualitativo. A maioria das anomalias relacionadas ao tipo 2A está inserida no éxon 28 do gene do FvW, no caso do tipo 2B também há concentração de anomalias no éxon 28, esses defeitos moleculares acontecem em regiões codificantes do domínio A1, onde está localizado o sítio de ligação com a glicoproteína Ib plaquetária. (GOODEVE, 2016).
O tipo 2M é em geral resultado de mutações posicionadas nos domínios A1 e D3, reduzindo sua afinidade com plaquetas, sem causar alterações a quantidade de multímeros de alto peso molecular. (STUFANO et al., 2015). Na DvW tipo 2N, as mutações, envolvendo os domínios D’ e D3, comumente se localizam entre os éxons 18 e 22, e decorrem em redução de afinidade do FvW pelo FVIII. (BOWMAN; JAMES, 2017).
A suspeita se inicia com um fenótipo de sangramento, mas devido a possibilidade da forma leve da doença, pode ocorrer a falta de sintomas em alguns pacientes, que só são diagnosticados se os exames forem realizados como parte da triagem genética caso um familiar seja portador da doença, porém alguns fatores reforçam a possibilidade de um diagnóstico clínico (quadro 2). A taxas sintomáticas da doença é muito mais baixa, estimada em aproximadamente 0,01% da população. (SWAMI; KAUR, 2017)
Quadro 2. Fatores reforçam a possibilidade de um diagnóstico clínico na DvW
| Condições mais comuns para diagnostico da DvW: |
| ● Histórico pessoal de sangramento |
| ● Histórico familiar positivo para sangramento e / ou diagnóstico de DvW |
| ● Testes laboratoriais que detectem anomalias qualitativas ou quantitativas no FvW |
Fonte: (BOWMAN; JAMES, 2017)
As ferramentas de avaliação de sangramento são uma parcela importante da observação diagnóstica, visto que podem ajudar a eleger os testes laboratoriais e padronizar a comunicabilidade dos históricos de sangramento entre os profissionais de saúde. Várias ferramentas de avaliação de sangramentos foram desenvolvidas e validadas. Ao longo dos anos, tem existido uma valorização progressiva sobre elas, para suprir esses contratempos diagnósticos. (DEFOREST et al., 2015).
Um estudo sobre ferramentas de avaliação de sangramento, evidenciou que ser do sexo masculino, facilitou a possibilidade de diagnostico em todas as avaliações. Levando em consideração que as aparições de sangramentos em homens sejam um sinal de alerta maior para os médicos e pacientes do que para mulheres, causando a descoberta e diagnostico mais rápido. (SPRADBROW et al., 2020).
Foi levantado também que 15% das mulheres com fluxo menstrual maior, possuam um transtorno hemorrágico hereditário subentendido, mais correntemente DvW, porém pela falta de distinção entre um sangramento menstrual normal e anormal e a mácula social sobre o tema, permanecem sem diagnostico. (SPRADBROW et al., 2020).
TESTES INICIAIS PARA DVW
Na rotina habitual de laboratório, o diagnóstico e a caracterização de cada tipo podem ser definidos por testes de laboratório, englobando um vasto painel de variados testes validados (figura 2). A aplicação clínica da presente categorização é comprometida por inúmeras alternâncias na reprodutibilidade e sensibilidade desses enxames diagnósticos; além do mais não leva em conta as modificações genéticas do FvW nem a estrutura do domínio da proteína. O diagnóstico pode ser complexo e nenhuma abordagem diagnóstica é adequada para todos os pacientes. (VANGENECHTEN et al., 2018).
Figura 2. Exames laboratoriais para diagnostico da DvW
|
Testes de triagem |
▪ Tempo de sangramento (TS)
▪ Tempo de trombloplastina parcial ativada (TTPA) ▪ Contagem plaquetária |
|
Testes confirmatórios |
▪ Atividade do fator VIII (FVIII:C)
▪ Antígeno do FvW (FvW:Ag) ▪ Atividade do co-fator de ristocetina (FvW:RCo) ▪ Capacidade de ligação do FvW ao colágeno (FvW:CB) |
|
Testes especiais* |
▪ Agregação plaquetária induzida pela ristocetina (RIPA)
▪ Capacidade de ligação ao FVIII (FVW:FVIIIB) ▪ Análise multimérica do FVW
|
Fonte: (PINHEIRO et al., 2017).
* Os testes especiais a facilitam a subclassificação da doença.
O diagnóstico laboratorial em pacientes pediátricos é complexo pela variação entre ensaios de FvW, aumentos instigados por estresse e carência de relatos de sangramento relevante com o qual confrontar os resultados dos exames. (DOSHI et al., 2019). Embora com testes concentrados, abrangendo ensaios funcionais a adição de uma ferramenta de avaliação de sangramento também não soluciona todas as complicações no diagnóstico. Não interfere onde a linha seja traçada para o diagnóstico da doença, o FvW é uma variante constante. Desse modo, pode ser uma desordem hemorrágica preocupante que demanda intervenção medicamentosa frequente ou uma condição leve que pode não ser clinicamente expressiva. (MONTGOMERY; FLOOD, 2016).
COMPLICAÇÕES HEMORRÁGICAS ASSOCIADAS A DVW EM PROCEDIMENTOS CIRÚRGICOS
O sangramento é uma complicação cirúrgica importante e está relacionada a taxas de mortalidade de 20% em episódios hemorrágicos. Cerca de 75% a 90% de sangramentos podem ser erros técnicos, porém coagulopatias podem facilitar ou causar diretamente hemorragia cirúrgicas em algumas situações. (BERNTORP et al., 2018). Um dano ao tecido é uma condição primária causadora da ruptura da continuação do endotélio, mediante circunstância acidental ou provocada. (SMILOWITZ et al., 2017).
Nos portadores da DvW, o sangramento é um episódio adverso que pode estar presente em procedimentos invasivos. Nessa compressão, até procedimentos invasivos de pequeno porte podem antecipar um incidente de sangramento demorado, que além de ser dolorido para o paciente, também impossibilita a finalização do procedimento e implica a recuperação da lesão. (LAINO et al., 2019)
Na literatura, a ocorrência de sangramentos pós operatório e após cirurgias na cavidade oral em pacientes com a hemostasia apropriada varia entre 0,2% e 3,3%, em conta partida a incidência de sangramentos pós operatório posteriormente cirurgia oral em portadores de desordens da hemostasia é estimada em 8,6% – 32,1%. Seja qual for o procedimento odontológico invasivo, expõe os pacientes com desordens da hemostasia a um grande risco de sangramento no intervalo pré e pós operatório. (DE PADUA et al., 2020).
As mulheres gravidas tem maior perigo a sangramentos se não tratadas, sangramento pré-parto, hemorragia pós-parto, hemorragia pós-parto grave e hematoma perineal aumentam de 2 a 10 vezes nas mulheres diagnosticadas. (JAMES et al., 2016).
As possibilidades de intervenções medicamentosas devem analisadas ao decorrer da gravidez, a condução da gestação é parcialmente fácil em portadoras do tipo 1 da doença, pois normalmente acontece uma elevação considerável nos níveis de FVIII e FvW. Entretanto, um acompanhamento rígido é sempre recomendável, uma vez que pode verificar-se um sangramento apesar da regularização desses fatores. (CASTAMAN; JAMES, 2019)
No caso da DvW tipo 2, seus subtipos devem ser seguidos com precaução, já que o aumento desarmônico dos fatores de coagulação ocorre relacionada a fisiopatologia de cada um. No tipo 3, clinicamente o mais grave, não há crescimento nos níveis de nenhum dos fatores, o que reivindica uma atenção maior no tratamento de reposição, que deve ser projetado com uma antecedência significativa. (CASTAMAN; JAMES, 2019).
Um estudo sobre do fator recombinante de von Willebrand em pacientes com doença grave de von Willebrand submetidos a cirurgia eletiva, citou como cirurgias que expressavam risco relevante de perda de uma quantidade significativa de sangue; cirurgias ortopédicas, abdominais, ginecológicas, de cabeça e pescoço, cirurgia intracraniana, cardiovascular ou espinhal. As práticas cirúrgicas menores compreenderam a introdução de dispositivos de acesso intravenoso, extração de pequenas lesões cutâneas, artroscopia, gastroscopia, colonoscopia ou conização. (MIESBACH, 2019).
TRATAMENTO DE SANGRAMENTO NO PERÍODO PERIOPERATÓRIO NA DVW
A DvW é definida especialmente por sangramento monocutâneo e após trauma ou cirurgia, o tratamento à disposição aplica-se a estabilização dos níveis de FvW e FVIII nesses casos. (DE JAGER et al., 2020). A classificação secundária da permite a intervenção adequada, o tratamento da hemóstase na DvW abrange o uso terapias não concentradas como pró-coagulantes, ácido aminocapróico, sendo o mais habitual o ácido tranexâmico, por via oral ou intravenosa, em sangramentos ou procedimento invasivos menores, e também a desmopressina utilizada para aumentar provisoriamente as taxas de FVIII e FvW. (LAFFAN et al., 2014).
No caso da terapia concentrada, a terapêutica de transfusão com produtos derivados de plasma FvW e FVIII da coagulação, mais indicado para hemorragias mais graves, intervenções cirúrgicas ou quando houver contraindicação ou não resposta ao uso de antifibrinolíticos e ao acetato de desmopressina. (FAVALORO et al., 2017).
O tratamento com os pró-coagulantes, em confronto com os concentrados do fator de coagulação, é uma escolha relativamente de baixo custo e possivelmente eficaz na prevenção de contratempos hemorrágicos em procedimentos na cavidade oral. Esses agentes não são indicados para pacientes com doença trombótica ativa. (VAN GALEN et al., 2015).
Atualmente entre as predominantes abordagens de tratamento disponíveis está a desmopressina, que é um análogo artificial da vasopressina e age promovendo a libertação das reservas endógenas do FvW dos corpos de WeIbel-Palade no endotélio vascular a afim de aperfeiçoar provisoriamente a homeostasia. Tendo em vista a versatilidade em feedback a esse medicamento em nível individual, sua efetividade nos diagnosticados precisa ser analisada com uma dose experimental antes de ser indicada para o uso na prevenção ou tratamento de episódios hemorrágicos. (MCCORMICK et al., 2018).
Ainda que possa ser sugerida como terapia de primeira escolha para cirurgias de pequeno porte e pacientes com o tipo 1, é limitada pela rápida diminuição do efeito farmacológico em doses consecutivas. Indagações com eventos adversos como hiponatremia, podem impossibilitar seu uso. Os hematologistas podem dar preferência a substituição do FvW através de concentrados em alguns casos clínicos, como em parto, cirurgia de grande porte, crianças com menos de 2 anos, pacientes com história de doenças cardiovasculares e pacientes com várias comorbidades associadas. (PEYVANDI et al., 2019).
Os concentrados de fator FvW/FVIII derivados do plasma, a princípio produzidos para o tratamento da hemofilia A, possuem quantidade elevada do FvW e são a terapêutica de escolha para os pacientes que não reagem ou que são intolerantes a desmopressina. (GILL et al., 2015). Contudo produtos oriundos do plasma não são tão abrangentes devido a disponibilidade de doadores de plasma e o perigo especulativo de propagação de patógenos não pode ser descartado, embora exista parâmetros de seleção e atenuação viral que foram criadas para reduzir preocupações como essa. (LEEBEEK; SUSEN, 2018).
A dosagem específica de produtos à base desses dois fatores para hemóstase em cirurgias, varia de acordo com a gravidade da doença e categoria da cirurgia. Inúmeras pesquisas prospectivas e retrospectivas explicitam as respostas hemostáticas desses concentrados em procedimentos cirúrgicos. Embora incomum, administrações seguidas, várias vezes no decorrer da cirurgia, foram relacionadas a episódios trombóticos associados a níveis elevados de atividade do FVIII. (SRIVASTAV et al., 2016).
Mesmo estando disponíveis há um tempo significativo, esses concentrados apresentam uma possibilidade teórica de contaminação, hipersensibilidade, reações alérgicas e trombose. (SINGAL; KOUIDES, 2016). O FvW elaborado por meio de técnicas recombinantes possibilita uma inovação na perspectiva do tratamento, excluindo as ameaças que podem estar relacionadas aos produtos vindos do plasma humano, assegurando a consistência e eficácia do produto. (MANNUCCI et al., 2013).
Os concentrados de fator recombinantes limitam o contato do paciente com FVIII, possibilitando que o profissional da saúde trate a alteração primária e especifique o tratamento baseado no fenótipo de sangramento. Com isso, possibilita uma dosagem mais repetida de FvW, se houver necessidade, sem o risco de acúmulo de níveis superiores ao normal. (HAZENDONK et al., 2018).
Cirurgias procedimento cirúrgicos podem ser executados com segurança, oferecendo a hemostasia apropriada e conveniente no decorrer e em seguida aos procedimentos nos portadores. Complicações hemorrágicas no perioperatório não são frequentes uma vez que os pacientes são monitorados com cautela. (ZULFIKAR et al., 2016).
TRATAMENTO POR TIPO DE DVW
No tipo 1, são indicados tratamentos que elevem de modo direto os níveis de FvW, como por exemplo o análogo sintético da vasopressina ou os concentrados, que habitualmente são indispensáveis apenas e para a reparação ou profilaxia de sangramentos severos, como acontece em traumas e procedimentos invasivos de grande porte. (FRANCHINI; MANNUCCI, 2016).
Os concentrados de fator de coagulação também são indicados em episódios hemorrágicos graves durante cirurgias no tipo 2A e 2B. A desmopressina deve ser testada antes do uso terapêutico, já que sua resposta é variável no primeiro caso e no segundo pode acelerar um agravamento de trombocitopenia. (TOSETTO; CASTAMAN, 2015).
Nos pacientes com tipo 2M e 3, o concentrado de FvW / FVIII é o tratamento de escolha. No tipo 2N a desmopressina pode ser administrada no tratamento de sangramentos menores, contudo o nível do FVIII é induzido a uma queda brusca, já que não é protegido pelo FvW, nessa perspectiva é necessário o concentrado de fatores para cobrir os procedimentos cirúrgicos. (WINDYGA et al.,2016).
CONSIDERAÇÕES FINAIS
A DvW pode ser fator de hemorragia grave com ameaça de vida, o perigo nos aumentos de sangramentos deve ser levado em consideração quando procedimentos médicos e eletivos são efetuados nesse distúrbio hemorrágico.
É de grande importância para os profissionais terem propriedade acerca do conceito, fisiopatologia e principalmente diagnósticos de coagulopatias hemorrágicas, visto que a semelhança das apresentações clínicas entre elas pode dificultar o diagnóstico, interferindo, na estipulação de um tratamento.
Através da análise da literatura, pode-se concluir que a desmopressina é ineficaz no tipo 3 e seu uso no tipo 2B permanece controverso devido à possibilidade de trombocitopenia. No entanto, ele pode ser usado efetivamente para cobrir pequenas cirurgias e procedimentos odontológicos na maioria dos outros pacientes. Para cirurgias de grande porte, há um uso mais amplo de concentrado de fator, garantindo que pacientes adequadamente tratados, passem por procedimentos invasivos sem grandes riscos de episódios hemorrágicos. O as opções terapêuticas para DvW se mostram eficazes para evitar o risco de sangramento intratável.
REFERÊNCIAIS
BRASIL. Ministério da Saúde. Secretaria de Atenção à Saúde. Departamento de Atenção Especializada. Manual de diagnóstico e tratamento da doença de von Willebrand. Ministério da Saúde, Secretaria de Atenção à Saúde, Departamento de Atenção Especializada. – Brasília: Editora do Ministério da Saúde, 2008.
BRASIL. Ministério da Saúde. Secretaria de Atenção à Saúde. Departamento de Atenção Especializada e Temática. Perfil das coagulopatias hereditárias: 2016 Ministério da Saúde, Secretaria de Atenção à Saúde. Departamento de Atenção Especializada e Temática. – Brasília: Ministério da Saúde, 2018. 57 p: il.
BERNTORP E, ÅGREN A, ALEDORT L, BLOMBÄCK M, CNOSSEN MH, CROTEAU SE, et al. Fifth Åland Island conference on von Willebrand disease. Haemophilia: the official journal of the World Federation of Hemophilia. 2018; vol. 24 Suppl 4: 5-19.
BOWMAN ML, JAMES PD. Controversies in the diagnosis of Type 1 von Willebrand disease. Int Jnl Lab Hem. 2017; 39(Suppl. 1): 61‐ 68.
CASTAMAN G, GOODEVE A, EIKENBOOM J, GRUO E, WILLEBRAND V. Principles of care for the diagnosis and treatment of von Willebrand disease. 2013;98(5):667–74.
CASTAMAN G, LINARI S. Diagnosis and Treatment of von Willebrand Disease and Rare Bleeding Disorders. J Clin Med. 2017;6(4):45.
CASTAMAN G, JAMES PD. Pregnancy and delivery in women with von Willebrand disease. Eur J Haematol. 2019; 103: 73– 79.
CAVALCANTI AJ, BRANDÃO JOC, LIMA MBPLV. Aspectos fisiopatológicos da doença de von Willebrand. Recife: Faculdade Integrada de Pernambuco – FACIPE, 2018. Trabalho de conclusão de curso em biomedicina.
DEFOREST M et al. Generation and optimization of the self-administered bleeding assessment tool and its validation as a screening test for von Willebrand disease. Haemophilia: the official journal of the World Federation of Hemophilia vol. 21,5 2015: e384-8.
DE JAGER NCB, BUKKEMS LH, HEIJDRA JM et al. OPTI-CLOT group. One piece of the puzzle: Population pharmacokinetics of FVIII during perioperative Haemate P®/Humate P® treatment in von Willebrand disease patients. J Thromb Haemost. 2020; 18: 295– 305.
DE PADUA V, UMBERTO RA, CRISTINA SB, RICCARDO BA, ERMINIA BALDACCI et al. Dental invasive procedures in von Willebrand disease outpatients treated with high purity FVIII/VWF complex concentrate (Fanhdi®): experience of a single center. Heliyon 2020; vol. 6,2 e03426.
DORUELO AL et al. Clinical and laboratory phenotype variability in type 2M von Willebrand disease. Journal of thrombosis and haemostasis: JTH vol. 15,8 2017: 1559-1566.
DOSHI BS, ROGERS RS, WHITWORTH HB, ET AL. Utility of repeat testing in the evaluation for von Willebrand disease in pediatric patients. J Thromb Haemost. 2019; 17: 1838– 1847.
ECHAHDI H, EL HASBAOUI B, EL KHORASSANI M, AGADR A, KHATTAB M. Von Willebrand disease: Case report and review of literature. Pan Afr Med J. 2017; 27:1–7.
EL OUAALITI M, LI R, GOBIN D, BRON D, CANTINIEAUX B. Diagnosis of congenital von Willebrand disease during a preoperative assessment in a multiple myeloma patient without bleeding history. Clin Case Rep. 2016;4(7):703-706.
FAVALORO EJ, SOMA M. Evaluation of a von Willebrand factor three test panel and chemiluminescent-based assay system for identification of, and therapy monitoring in, von Willebrand disease. Thrombosis research vol. 141 2016: 202-11.
FAVALORO EJ et al. Monitoring Therapy during Treatment of von Willebrand Disease. Seminars in thrombosis and hemostasis vol. 43,3 2017: 338-354.
FRANCHINI M, MANNUCCI PM. Von Willebrand factor (Vonvendi®): the first recombinant product licensed for the treatment of von Willebrand disease. Expert review of hematology vol. 9,9; 2016: 825-30.
GILL JC, CASTAMAN G, WINDYGA J, KOUIDES P, RAGNI M, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood vol. 126,17; 2015: 2038-46.
GOODEVE A. Diagnosing von Willebrand disease: genetic analysis. Hematology. American Society of Hematology. Education Program vol. 2016,1 2016: 678-682.
GUERRERO B, LÓPEZ M. Generalidades del sistema de la coagulación y pruebas para su estudio. Invest. clín. 2015; 56(4): 432-454.
HAZENDONK HCAM, HEIJDRA JM, DE JAGER NCB, VEERMAN HC, BOENDER J et al. Análise do manejo perioperatório atual com Haemate ® P / Humate P ® na doença de von Willebrand: identificando a necessidade de tratamento personalizado. Hemofilia. 2018; 24: 460 – 470.
HEIJDRA JM, CNOSSEN MH, LEEBEEK FWG. Current and Emerging Options for the Management of Inherited von Willebrand Disease. Drugs. 2017;77(14):1531–47.
JAMES AH, EIKENBOOM J, FEDERICI B. State of the art: von Willebrand disease. Haemophilia. 2016; 22: 54-59.
JAMES PD, GOODEVE AC. Von Willebrand disease. Genetics in medicine: official journal of the American College of Medical Genetics vol. 13,5 (2011): 365-76.
KEESLER DA, FLOOD VH. Current issues in diagnosis and treatment of von Willebrand disease. Research and practice in thrombosis and haemostasis vol. 2,1 34-41. 12 Dec. 2017.
LAFFAN MA, LESTER W, O’DONNELL JS, WILL A, TAIT RC, GOODEVE A, et al. The diagnosis and management of von W illebrand disease: a U nited K ingdom Haemophilia C entre Doctors Organization guideline approved by the B ritish Committee for Standards in Haematology. Br J Haematol. 2014; 167: 453-465.
LAINO L, CICCIÙ M, FIORILLO L, CRIMI S, BIANCHI A.; AMOROSO G et al. Surgical Risk on Patients with Coagulopathies: Guidelines on Hemophiliac Patients for Oro-Maxillofacial Surgery. International journal of environmental research and public health. 2019; vol. 16,8 1386.
LAVIN M et al. Novel insights into the clinical phenotype and pathophysiology underlying low VWF levels. Blood vol. 130,21 2017: 2344-2353.
LAVIN M, O’DONNELL JS. New treatment approaches to von Willebrand disease. Hematology Am Soc Hematol Educ Program. 2016;2016(1):683–689.
LEEBEEK FWG, FERDOWS A. How I manage severe von Willebrand disease. British journal of haematology. 2019 vol. 187,4; 418-430.
LEEBEEK FWG, SUSEN S. Von Willebrand disease: Clinical conundrums. Haemophilia. 2018; 24(Suppl. 6): 37‐ 43.
LENTING PJ, CHRISTOPHE OD, DENIS CV. Von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood 2015; vol. 125,13: 2019-28.
MANNUCCI PM, KEMPTON C, MILLAR C, ROMOND E, SHAPIRO A, BIRSCHMANN I et al. Pharmacokinetics and safety of a novel recombinant human von Willebrand factor manufactured with a plasma-free method: a prospective clinical trial. Blood vol. 122,5 2013: 648-57.
MCCORMICK MC, SIRIPONG N, COOPER JD. Desmopressin stimulation testing: Response to intravenous and intranasal forms. Haemophilia. 2018; 24: e194– e198.
MIESBACH W. Phase 3 study of recombinant von Willebrand factor in patients with severe von Willebrand disease who are undergoing elective surgery: Comment. Journal of thrombosis and haemostasis: JTH. 2019; vol. 17,8: 1403-1405.
MONTGOMERY R, FLOOD VH. What have we learned from large population studies of von Willebrand disease? Hematology Am Soc Hematol Educ Program 2016; 2016 (1): 670–677.
NG C, MOTTO DG, DI PAOLA J. Diagnostic approach to von Willebrand disease. Blood. 2015;125(13):2029–2037.
PEYVANDI F, PETER KOUIDES, TURECEK PL, DOW E, BERNTORP E et al. Evolution of replacement therapy for von Willebrand disease: From plasma fraction to recombinant von Willebrand factor. Blood reviews vol. 38 2019: 100572.
PINHEIRO YT, DA SILVA ECL, MACIEL MA, DE SOUSA et al. Hemofilias e Doença de von Willebrand: uma revisão de literatura. Arch Heal Investig. 2017;6(5).
RUGGERI ZM, MENDOLICCHIO GL. Interaction of von Willebrand factor with platelets and the vessel wall. Hamostaseologie vol. 35,3 2015: 211-24.
SANTOS, AVO. Uso do DDAVP e do concentrado de CFvW/FVIII em pacientes com doença de Von Willebrand do Hemocentro de Belo Horizonte entre 2011 e 2013. Ribeirão Preto: Universidade de São Paulo, Faculdade de Medicina de Ribeirão Preto; 2017.
SINGAL M, KOUIDES PA. Recombinant von Willebrand factor: a first-of-its-kind product for von Willebrand disease. Drugs Today (Barc). 2016;52(12):653-664.
SMILOWITZ NR, GUPT, N, GUO Y et al. Perioperative bleeding and thrombotic risks in patients with Von Willebrand disease. J Thromb Thrombolysis. 2017; 44, 67–70.
SPRADBROW J, LETOURNEAU S, GRABELL J, et al. Bleeding assessment tools to predict von Willebrand disease: Utility of individual bleeding symptoms. Res Pract Thromb Haemost. 2020; 4: 92– 99.
SRIVASTAV A, SERBAN M, WERNER S, SCHWARTZ BA, KESSLER CM. Efficacy and safety of a VWF /FVIII concentrate (wilate®) in inherited von Willebrand disease patients undergoing surgical procedures. Haemophilia, 2016; 23: 264-272.
STUFANO F, BARONCIANI L, PAGLIARI MT, FRANCHI F, COZZI G, GARCIA‐OYA I, BUCCIARELLI P, BOSCARINO M, PEYVANDI F. Evaluation of an heterogeneous group of patients with von Willebrand disease using an assay alternative to ristocetin induced platelet agglutination. J Thromb Haemost 2015; 13: 1806– 14.
SWAMI A, KAUR V. Von Willebrand Disease: A Concise Review and Update for the Practicing Physician. Clin Appl Thromb. 2017;23(8):900–10.
VAN DIEVOET M-A, EECKHOUDT S, STÉPHENNE X. Primary Hemostasis in Chronic Liver Disease and Cirrhosis: What Did We Learn over the Past Decade? International journal of molecular sciences. 2020; vol. 21,9 E3294.
VAN GALEN KPM, ENGELEN ET, MAUSER‐BUNSCHOTEN EP, VAN ES RJJ, SCHUTGENS REG. Antifibrinolytic therapy for preventing oral bleeding in patients with haemophilia or Von Willebrand disease undergoing minor oral surgery or dental extractions. The Cochrane database of systematic reviews ,12 CD011385. 24 Dec. 2015.
VANGENECHTEN I, MAYGER K, SMEJKAL P, ZAPLETAL O, MICHIELS JJ, MOORE GW, GADISSEUR A. A comparative analysis of different automated von Willebrand factor glycoprotein Ib‐binding activity assays in well typed von Willebrand disease patients. J Thromb Haemost 2018; 16: 1268– 77.
TOSETTO A, CASTAMAN G. How I treat type 2 variant forms of von Willebrand disease. Blood 2015; 125 (6): 907–914.
WINDYGA J, DOLAN G, ALTISENT C, KATSAROU O, LÓPEZ FMF, ZÜLFIKAR B. Practical aspects of DDAVP use in patients with von Willebrand Disease undergoing invasive procedures: a European survey. Haemophilia, 2016; 22: 110-120.
ZULFIKAR B; KOC B, GULSUM AK; DIKICI F, KARAMAN İHSAN, ATALAR ATA CAN; BEZGAL FIKRET. Cirurgia em pacientes com doença de Willebrand, Coagulação sanguínea e fibrina: outubro de 2016 – Volume 27 – Edição 7 – p 812-816. (ZULFIKAR et al., 2016).
[1] Discente do curso de graduação em farmácia do Centro Universitário Maurício de Nassau, Recife, Pernambuco, Brasil.
[2] Farmacêutico, Responsável Técnico da Drogaria Mais Você, Pós Graduando em Farmácia Clínica com Atenção Farmacêutica pela FARESE.
[3] Farmacêutico Generalista.
[4] Farmacêutico Generalista.
[5] Orientador. Docente do Centro Universitário Maurício de Nassau. Especialista em Citologia Clínica. Citopatologista responsável pelo Controle de Qualidade Interno do Laboratório HEMOLAB diagnósticos.
Enviado: Setembro, 2020.
Aprovado: Setembro, 2020.