TAVARES, Andressa Santos [1], PICHATELLI, Fernanda Pagnan [2], PRADO, Marnie Chaves Genari Brandao [3]
TAVARES, Andressa Santos; et.al. A Importância da Validação de Limpeza na Indústria Farmacêutica: Revisão de Literatura. Revista Científica Multidisciplinar Núcleo do Conhecimento. Ano 03, Ed. 01, Vol. 02, pp. 85-100, Janeiro de 2018. ISSN: 2448-0959
RESUMO
Baseado nas boas práticas de fabricação (BPF), a validação de limpeza é um método utilizado para assegurar que os procedimentos de limpeza de equipamentos sejam efetuados de maneira que todos os resíduos sejam eliminados até um determinado nível de aceitação. Por isso a validação é de extrema importância na indústria farmacêutica para garantir a remoção de resíduos de produtos recém-fabricados, não afetando a qualidade dos processos e produtos produzidos. O presente trabalho tem por objetivo enfatizar a seriedade da validação de limpeza nos processos de produção dos medicamentos. O método de validação de limpeza mais utilizado consiste em desmontar todo o equipamento, lavar com água potável, detergente neutro e lavagem com água purificada, posteriormente efetuando a amostragem. A validação da metodologia analítica é necessária para garantir que a quantificação de resíduos condiz com os critérios adotados na validação de limpeza. O método mais comum utilizado é a amostragem por SWAB, utilizada para verificar presença de resíduos em partes dos equipamentos que são consideradas superfícies difíceis de limpar, superfícies essas determinadas na validação do processo. Após a amostragem, são feitos testes de controle de qualidade para garantir a total limpeza dos equipamentos e evitar contaminações cruzadas de carga microbiana. Verifica-se que com o emprego das BPF é possível garantir a qualidade dos procedimentos de limpeza e assim dos produtos fabricados pela indústria, sendo necessário o alinhamento da indústria com a garantia da qualidade para diminuir os riscos e consequentemente a ocorrência de relatos de casos de contaminação cruzada.
Palavras-Chave: Validação de Limpeza, Método, Medicamentos.
INTRODUÇÃO
Um dos maiores desafios que as indústrias farmacêuticas enfrentam é a contaminação, todos os produtos farmacêuticos e ativos ou insumos que compõem uma formulação podem ser contaminados e essa contaminação pode ocorrer por agentes de limpeza, solvente, por produtos degradados, microrganismos ou outros materiais. Para evitar todos esses tipos de contaminação os processos de limpeza foram criados e devem ser robustos e validados, garantindo assim a segurança do medicamento produzido (LAURO D’MORETTO, 2016).
Na indústria o setor responsável pela Validação de Limpeza, é a Garantia da Qualidade, esse setor deve cobrir todos os assuntos e aspectos que influenciam individualmente ou coletivamente na qualidade do produto. A garantia é a responsável por validar todos os processos necessários para a fabricação de um medicamento, inclusive a validação de limpeza, a principal missão da garantia é assegurar que o medicamento não seja comercializado ou distribuído antes que os responsáveis de cada área se certifiquem que a cada lote produzido foi controlado de acordo com o registro e quaisquer outras normas relevantes ao processo de produção, isso envolve conferencia da ordem de produção e dos documentos de validação, relacionados ao lote produzido (BRASIL, 2013).
Validar um processo significa garantir e documentar que aquele processo está sendo feito da mesma forma, de acordo com a especificação, limites de tolerância e normativas dispostas. O termo validar dentro da indústria farmacêutica assegura que aquela atividade ou aquele processo já foi testado e documentado, de forma que deve ser seguido até que uma mudança, ou um problema seja detectado (BRASIL, 2006).
As boas práticas de fabricação é a parte da Garantia a Qualidade que auxilia na segurança de que todos os produtos são produzidos e controlados com os padrões de qualidade apropriados para o uso pretendido e o requerimento do registro, essas normas orientam na diminuição de riscos durante a produção de qualquer produto farmacêutico, esses riscos podem ser por contaminação cruzada, contaminação de partículas e a troca ou a mistura de produtos, que podem ocorrer durante a pesagem dos insumos (BRASIL, 2013).
A história da indústria farmacêutica no Brasil e em outros países apresenta mais alguns episódios de contaminação cruzada envolvendo a produção e a qualidade de produtos farmacêuticos, são os casos do contraceptivo oral o Microvlar® e o medicamento Celobar®, que é um contraste na época usado em exames radiológico, esse caso levou ao óbito cerca de 20 pacientes em Goiânia, que foram intoxicados por carbonato de bário (BaCO3), composto utilizado para a síntese do contraste que não deveria estar no final da reação por ser tóxico (TUBINO e SIMONI, 2007).
A produção de medicamentos envolve, além de processos físicos, processos químicos complexos, podendo assim levar a produção de uma infinidade de subprodutos, inclusive tóxicos. Durante os processos, são utilizados vários reagentes e solventes, essas substâncias devem ser eliminadas e não devem ser detectadas no produto final, sendo então impossíveis de permanecer em equipamentos ou utensílios, contaminando outros lotes de produtos do mesmo medicamento ou de outros que possam vir a ser produzidos em seguida (HEALTH PRODUCTS, 2000).
O cenário de repentinos casos de contaminação cruzada levou a um índice significativo de reprovações no exercício da validação de limpeza, transformando-a num processo moroso, complexo e de alto custo, devido aos investimentos em pessoas e outros recursos necessários para o cumprimento dos planos de execução. Para se evitar tais reprovações na validação os processos de limpeza se tornaram mais robustos, resultam assim em maior gasto de energia, agentes de limpeza, água, geração de efluentes e reduzindo a capacidade produtiva dos equipamentos (LAURO D’MORETTO, 2016).
A validação de limpeza garante que os resíduos remanescentes do lote produtivo estejam sendo retirados, a níveis aceitáveis, durante o processo de limpeza. Observa-se, através de monitoramento analítico, a remoção dos produtos residuais, produtos de degradação, conservantes, excipientes e/ou agentes de limpeza de modo a obedecer às especificações e limites de cada um desses. Além disso, é necessário garantir a inexistência de riscos associados com contaminação cruzada de princípios ativos (HEALTH PRODUCTS, 2000).
Uma análise de risco é realizada para que através de um levantamento sejam determinadas as semelhanças físicas dos produtos, é feito também a análise das formulações, a quantidade e a forma que essa formulação será utilizada pelo consumidor final, os equipamentos que são compartilhados entre um produto e outro e o tamanho do lote. A escolha do ‘’Pior Caso’’ é feita de acordo com a denominação de todos os produtos contaminantes, através de procedimentos analíticos que evidenciará que após a limpeza não haverá resíduo de outras substâncias na linha de processo que possa acarretar em uma contaminação cruzada. Para determinar um representante entre um portfólio variado de produtos, é aceitável que o mesmo apresente características críticas em relação aos outros (BRASIL, 2006; FDA, 1993).
Um procedimento de limpeza adequado deve ser desenvolvido para todos os equipamentos e utensílios que vão entrar em contato com o produto durante o processo de produção, em um processo de avaliação pela Validação de Limpeza, temos que avistar a eficácia dos procedimentos de limpeza. As peças dos equipamentos também devem ser consideradas, elas podem não entrar em contato direto com o produto, porém dificultam e podem comprometer a limpeza. As peças são consideradas críticas, pois dificultam o acesso ao local da limpeza ou áreas que não tenham contato com o produto, enfatizando então, que todo o equipamento deve estar limpo (FDA, 1993, HEALTH PRODUCTS, 2000).
Existem alguns equipamentos que exigem que a limpeza seja feita, como por exemplo, os filtros de leitos fluidizados, produtos com alta atividade biológica ou toxicidade. Em casos de campanhas, onde são realizadas limpezas parciais entre diferentes lotes de produção, a validação de limpeza deve definir o tempo máximo da campanha. No estudo, deve ser considerada também a pesquisa das impurezas (BRASIL, 2006).
O presente trabalho tem como objetivo realizar uma revisão bibliográfica sobre a importância da validação de limpeza na indústria farmacêutica, que é uma exigência para a obtenção da Certificação de Boas Práticas de Fabricação com ênfase nas características e pontos críticos para a escolha de um produto como pior caso. A validação é um assunto pouco abordado, porém de grande importância na indústria farmacêutica, por isso a necessidade de ressaltar esse assunto.
Através das bases das literaturas, podemos analisar os parâmetros que devem ser seguidos para se efetivar um processo de validação de limpeza de acordo com as legislações vigentes.
METODOLOGIA
A metodologia utilizada para atingir o objetivo deste trabalho foi elaborada através de uma revisão bibliográfica abrangendo as principais legislações atuais vigentes sobre validação de limpeza. São essas a RDC 17 que dispõe sobre as Boas Práticas de Fabricação, a RDC 249 que fala do Regulamento Técnico das Boas Práticas de Fabricação de Produtos Farmacêuticos Ativos, a RDC 69 que dispõe sobre as Boas Práticas de Fabricação de Insumos Farmacêuticos Ativos, Farmacopéia Brasileira 5ª edição, Guia de Validação de Limpeza para Farmoquímicas da ANVISA, Guias Relacionados à Garantia da Qualidade da ANVISA, livro publicado pela SINDUSFARMA sobre Qualificação e Validações: Guia Sindusfarma para Indústria Farmacêutica e também artigos científicos publicados, sendo pesquisados basicamente através das ferramentas de buscas eletrônicas, da Web, como Goolge Acadêmico e Scientific Electronic Library Online (SciELO).
Todo o material foi pesquisado no período de maio, junho, julho, agosto e setembro de 2017, tendo também como apoio ao conhecimento do assunto a Indústria farmacêutica Cristália, que atua a mais de 45 anos na fabricação de medicamentos e ativos farmacêuticos, onde atualmente ambas as autoras cumprem o estágio.
RESULTADOS E DISCUSSÃO
A atenção e a preocupação com a qualidade e segurança na produção farmacêutica aumentaram devido aos casos ocorridos na década de 60, como o aparecimento de características sexuais secundárias em crianças que teriam sido relatados como consequência da contaminação cruzada no medicamento isodiazina (hidrazina do ácido isonicotínico com dietilestilbestrol) (COUTINHO et al., 2009).
Um surto de salmonelose foi mais um caso onde a contaminação cruzada interferiu no tratamento de doentes, um comprimido usado por cerca de 240 pessoas para tratamento de hipertireodismo, cinco delas foram contaminadas por dois tipos diferentes de salmonelas, a S. muenchen e a S. bareilly. Realizadas as investigações verificou-se que a contaminação havia sido pela matéria prima utilizada na fabricação dos comprimidos, na qual havia uma quantidade de bactérias acima do permitido, sendo a maior parte da flora fecal (KAWANO et. al., 2006).
Os excipientes, também podem apresentar riscos na produção de medicamentos, como no caso ocorrido no Haiti, onde foram a óbito 88 crianças por insuficiência renal aguda após a ingestão de xarope de paracetamol devido à glicerina utilizada na preparação contaminada com dietilenoglicol (O’BRIEN ET AL., 1998). De acordo com excipientes são substâncias adicionadas à formulação com atividade específica par atuar como aglutinantes, desintegrante, diluente entre outros, por isso deve ser analisada assim como os medicamentos sendo submetidos ao controle de qualidade. (PRISTA, 2002 apud RAMOS e MORAIS, 2013).
Os insumos farmacêuticos ativos (IFA) tem propriedade farmacológica com finalidade medicamentosa. Por ser o princípio ativo do medicamento o seu controle de qualidade também deve ser rigoroso (ANVISA, 2016).
Outros casos de contaminação cruzada com princípios ativos ou substâncias químicas tiveram como consequência à retirada de vários produtos farmacêuticos do mercado, fatos que deram origem a um problema de saúde pública. Portanto, tornou-se necessária a criação de medidas preventivas e corretivas para tal problema (COUTINHO et al., 2009).
Segundo a ANVISA, 2010 as áreas e equipamentos das indústrias farmacêuticas podem ser compartilhados para os produtos produzidos, desde que a mesma mantenha procedimentos de limpeza ou inativação validados, com exceção de alguns insumos como cefalosporinas, penicilinas, carbapenêmicos e outros derivados betalactâmicos, que necessitam de áreas dedicadas para sua produção devido a sua característica altamente sensibilizante. As indústrias de medicamentos realizam a validação dos processos de limpeza frente à possibilidade de contaminação cruzada, a fim de buscar a segurança dos usuários dos medicamentos e também para atender as determinações regulatórias vigentes de Boas práticas de Fabricação (BPF) (ANVISA, 2010).
Para se iniciar um estudo de Validação de limpeza, primeiramente devem ser mapeadas todas as rotas dos produtos existentes na fábrica, assim podemos determinar o agrupamento dos equipamentos que cada setor possui, dando origem as rotas de produção onde vão ser evidenciados os piores casos e as metodologias a serem utilizadas, para documentar a eficácia do processo de limpeza a ser comprovado. Os dados toxicológicos devem ser comprovados com os limites de aceitação encontrados, assim será montada uma matriz de pior caso (BRASIL, 2006).
Tais procedimentos devem ser documentados assegurando que a limpeza é capaz de remover os resíduos de produtos e agentes de limpeza para assim evitar a contaminação de lotes produzidos posteriormente, documentos estes que são assegurados pelo setor da Garantia de Qualidade. Para isso, o método de amostragem e os métodos analíticos a serem usados para realização da avaliação da limpeza, também devem ser validados e devem ter a sensibilidade para detectar resíduos ou contaminantes (BRASIL, 2010).
O setor de Controle de qualidade por sua vez, fica responsável por realizar as análises emitir os resultados documentados para enviar à Garantia da Qualidade. A realização do controle de qualidade é de extrema importância na indústria farmacêutica para assegurar a eficácia, segurança e confiabilidade dos produtos. (ROCHA; GALENDE, 2014).
Depois de todo o processo de limpeza deve ser feita uma etiqueta de status de limpeza, nessa etiqueta deve constar o nome, concentração, lote do último produto utilizado no equipamento ou utensílio, prazo de validade da limpeza e nome dos funcionários que executaram e supervisionaram a limpeza, essa etiqueta é de extrema importância para caso ocorra uma contaminação cruzada, seja possível investigar todo o processo (BRASIL, 2006).
Os protocolos de Recuperação de Ativo devem ser definidos e determinados quais serão os critérios de aceitação a serem adotados para os resíduos químicos e resíduos de agentes de limpeza que serão admitidos. A contaminação microbiológica é outro tipo de contaminação com um potencial para adulterar a qualidade do medicamento. Essa forma de contaminação, mesmo que a limpeza tenha sido feita, muitas vezes surpreende podendo ocorrer de forma eficaz (BRASIL, 2006).
Os equipamentos não dedicados têm por necessidade uma grande quantidade de procedimentos de limpeza, por isso podem ser aplicados diferentes níveis de limpeza, como mostra a Tabela I, com a finalidade de reduzir o número de procedimentos necessários, mantendo a segurança do IFA ou medicamento (ANVISA, 2010).
Tabela 1 – Níveis de Limpeza
| Nível | Abrangência | Validação de limpeza |
| 2 | Substâncias passíveis de serem transferidas do lote anterior são críticas, dessa forma a limpeza é necessária até que o limite determinado da quantidade transferida seja alcançado. | Essencial |
| 1 | Substâncias passíveis de serem transferidas do lote anterior são menos críticas sendo que a limpeza deve reduzir a quantidade potencial transferida para um limite menos severo do que necessário para o nível 2. | De não obrigatória a necessária (limites menores de produtos transferidos aceitáveis) |
| 0 | Somente limpeza bruta, se as substâncias passíveis de serem transferidas do lote anterior não são críticas. | Não necessária |
Fonte: Guia de validação de limpeza para Farmoquímicas.
Para a validação de limpeza dos equipamentos deve ser estabelecido por quanto tempo o equipamento pode ficar sujo, antes que a limpeza seja executada, pois a efetividade de um procedimento de limpeza é inversamente proporcional ao tempo que o mesmo permanece sujo, sobretudo para os produtos tópicos, suspensões, formulações com gelatina, onde a secagem do resíduo aumenta consideravelmente a dificuldade de limpeza. Caso seja definido que o equipamento pode permanecer sujo por 24 horas antes que seja executada a limpeza, então o estudo de validação e a limpeza só poderão ser conduzidos nesse prazo limite para assegurar que o procedimento do pior caso seja seguro (BRASIL, 2006).
Para validação do processo de limpeza, são amostrados pontos do equipamento pré-determinados, escolhendo os locais mais críticos de limpeza que devem ser indicados no protocolo de validação, geralmente são locais de difícil acesso para limpeza, ranhuras e locais apontados pelos operadores como mais difíceis de realizar a limpeza. São realizadas as análises de riscos para indicar um produto como “pior caso” e os critérios de aceitação, conforme indicado na Figura 1. A análise de risco leva em consideração vários fatores, como a classe terapêutica do medicamento, a dificuldade de limpeza, produto secundário ou produtos de degradação, toxicidade, solubilidade do princípio ativo, menor e maior dose terapêutica, número mínimo de lote dentre outros parâmetros. Os resultados das análises após a limpeza devem cumprir as especificações pré-determinadas de forma que demonstre eficiência para evitar a contaminação cruzada (BRASIL, 2010).
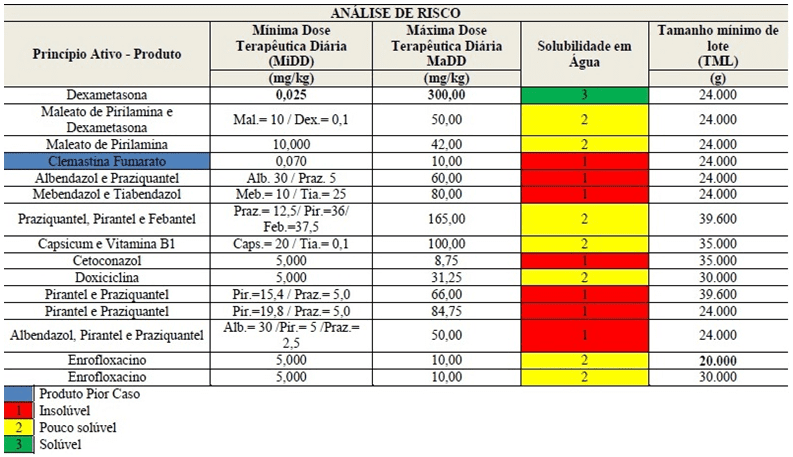
Para cada tipo de equipamento e medicamentos produzidos, o procedimento também deve conter detalhes sobre o tempo que o equipamento pode permanecer sujo até a limpeza, pois cada vez mais seco mais difícil de ser removido, determinar a quantidade de solvente a ser utilizado, o tipo e a concentração do detergente e os métodos empregados na limpeza (ANVISA, 2006).
Em indústrias que contém produção em grande escala, faz-se necessário a validação de lotes em campanha, um estudo de extrema importância para definir a quantidade máxima de lotes produzidos em campanha sem a necessidade de realizar uma limpeza completa dos equipamentos envolvidos no processo entre a produção de um lote e outro. Conforme descrito na RDC n° 17/10 de Boas Práticas de Fabricação de Medicamentos, deve-se evitar a contaminação cruzada e contaminação microbiana durante a produção por meio de técnicas validadas de limpeza, determinando a quantidade de lotes que podem ser produzidos sem a realização de limpeza completa dos equipamentos. Da mesma forma, é específica na RDC N° 69/14 de Boas Práticas de Fabricação de Insumos Farmacêuticos Ativos na Seção V – Controle de contaminação no Art. 187°. A periodicidade da limpeza irá garantir que os materiais residuais que possam ser emanados do produto produzido para o produto subsequente, não alterem a qualidade do produto (BRASIL, 2010; BRASIL, 2014).
Conforme as mudanças nos processos de produção, de equipamentos, métodos analíticos, ocorrência de contaminação, alterações intencionais ou não, em certos casos é necessário realizar a revalidação para assegurar que tais alterações não afetaram a qualidade do medicamento ou insumo farmacêutico ativo. Através do acompanhamento do processo de controle de mudanças estabelecido pela empresa é possível avaliar a necessidade de revalidação (BRASIL, 2010).
Como medidas preventivas a fim de minimizar os riscos de contaminação cruzada ou microbiológica, as resoluções vigentes devem ser seguidas e atualizadas conforme as necessidades. As auditorias internas e externas têm se tornando cada vez mais rigorosas quando se trata dos assuntos de validação de limpeza e controle de qualidade. Por isso, a resolução dispõe que todas as etapas dos processos industriais devem ser devidamente definidas, validadas e regularmente revisadas e sempre documentadas. Incluindo a infraestrutura da empresa, que também deve seguir um modelo apropriado, pessoal qualificado e treinado para os procedimentos envolvidos na produção, os equipamentos com qualificações e manutenções atualizadas, armazenamento e transporte adequados, além de estrutura para controle de processos (BRASIL, 2010).
Com o aumento da variedade de produtos produzidos na indústria farmacêutica, para aperfeiçoar o processo e diminuir os gastos, passou-se então a se admitir a escolha do ‘Pior Caso’ para representar a limpeza de todos os equipamentos, que compõe sua rota de produção, assim ao aprovar o ‘‘Pior Caso’’ na validação de limpeza automaticamente estará assumindo que todo o produto fabricado naquele equipamento tem seu processo validado (BRASIL, 2006).
No Brasil requerimentos foram criados para aumentar o rigor das ações referentes à contaminação cruzada, são eles os mais importantes RDC nº 17 de 16 de Abril de 2010 que dispõe sobre as Boas Práticas de Fabricação e a RDC Nº 69 DE 8 de Dezembro de 2014 que dispõe sobre as Boas Práticas de Fabricação de Insumos Farmacêuticos Ativos, a criação desses requerimentos foi necessária para diminuir os casos de contaminação cruzada, elas enfatizam que todos os equipamentos devem ser limpos de acordo com um POP (Procedimento operacional Padrão), levando em consideração todos os aspectos referente a validação (BRASIL, 2010, BRASIL, 2013).
Nos dias de hoje, devido à grande variedade de produtos no portfólio das indústrias farmacêuticas, existe a dificuldade em se dedicar a uma unidade de fabricação ou no conjunto de equipamentos do processo produtivo de um único produto. É necessário garantir a segurança de que o lote/produto subseqüente não receba nenhum resíduo tanto do produto anterior quanto o de produtos utilizados durante o processo de limpeza dos equipamentos (HEALTH PRODUCTS, 2000).
CONCLUSÃO
Inúmeros são os riscos de contaminação cruzada e microbiana em insumos e produtos farmacêuticos, por isso o aumento constante de exigências pela vigilância sanitária. O alinhamento da indústria com As Boas Práticas de Fabricação (BPF) é necessário para diminuir os riscos e consequentemente a ocorrência de relatos de casos de contaminação cruzada.
Pelos estudos realizados, verifica-se que com o emprego das BPF é possível verificar e garantir a qualidade dos procedimentos de limpeza e assim dos produtos fabricados pela indústria. A análise de risco e escolha do pior caso durante a validação de limpeza contribui para a produção de medicamentos seguros e auxilia na eliminação de uma possível contaminação. Assim, é possível identificar e corrigir problemas de qualidade ocorridos ou evitá-los. A validação documentada é de extrema importância para assegurar que os testes estão sendo executados de forma correta e confiável conforme especificado em seus procedimentos.
O crescimento industrial pode ser obtido através do gerenciamento desses riscos, por diminuir o número de desvios de qualidade e assegurar confiabilidade aos seus produtos. Porém, esses procedimentos e etapas têm como objetivo atender as especificações para proporcionar aos pacientes o uso de medicamentos adequados e livres de contaminação que possam afetar a sua saúde.
REFERÊNCIAS
Americanas em Ciências, Maringá, p.26-31, 22 abr. 2007. Disponível em: <http://xa.yimg.com/kq/groups/29904052/365417193/name/ArtigoValidaçãoMetodologiaAnalítica.pdf.>. Acesso em: 08 mai. 2017.
ANVISA. Guia de Validação de limpeza para Farmoquímicas. Disponível em: <http://portal.anvisa.gov.br/documents/33836/2501339/Guia+de+valida%C3%A7%C3%A3o+de+limpeza+para+farmoqu%C3%ADmicas/573873c0-f41c-42bd-833f-d01f5767cebd> Acesso em: 08 mai. 2017.
BRASIL, Agência Nacional de Vigilância Sanitária, farmacopéia Brasileira 5° ed., Vol.2, Brasília, DF, 2010e.
BRASIL. Resolução nº 17, de 16 de novembro de 2010. Resolução RDC Nº 17, de 16 de Abril de 2010. 1. ed. Brasil. Disponível em: <http://portal.anvisa.gov.br/documents/33880/2568070/res0017_16_04_2010.pdf/b9a8a293-f04c-45d1-ad4c-19e3e8bee9fa> Acesso em: 22 mai. 2017.
BRASIL, Ministério da Saúde, ANVISA, Guias relacionados à garantia de qualidade de 31 de outubro de 2006. Disponível em: < www.anvisa.gov.br> Acesso em: 06 de set. de 2017.
BRASIL, Ministério da Saúde, ANVISA. Resolução nº 249 de 13 de setembro de 2005. Regulamento Técnico das Boas Práticas de Fabricação de Produtos Intermediários e Insumos Farmacêuticos Ativos.
COUTINHO, Roberto C. et al. Determinação simultânea de resíduos de sulfametoxazol e trimetoprima em superfícies de equipamentos de produção. Química Nova, [s.l.], v. 32, n. 8, p.2214-2217, 2009. Fap UNIFESP (SciELO). Disponível em: <http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0100-40422009000800038>. Acesso em: 16 set. 2017.
HEALTH PRODUCTS AND FOOD BRANCH INSPECTORATE. CleaningValidation Guidelines 2000. Disponível em: <https://www.canada.ca/content/dam/hc-sc/migration/hc-sc/dhp-mps/alt_formats/hpfb-dgpsa/pdf/compli-conform/gui_0028_tc-tm-eng.pdf> Acesso em: 07 de set. de 2017.
KAWANO, D. F. Acidentes com os medicamentos: como minimizá-los? Revista Brasileira de Ciências Farmacêuticas (Brazilian Journal of Pharmaceutical Sciences) vol. 42, n. 4, out./dez., 2006.
LA ROCA, Mônica Felts de, et al. Desenvolvimento e validação de método analítico: passo importante na produção de medicamentos. Revista Brasileira Farmacêutica, Recife/pe, v. 4, n. 88, p.177-180, 10 jul. 2007. Disponível em: <http://www.rbfarma.org.br/files/pag_177a180.pdf>. Acesso em: 22 mai. 2017.
LAURO D’MORETTO (São Paulo). Sindusfarma. Guia de Qualificação e validação. 17. ed. São Paulo: Câmara Brasileira do Livro, 2016.
MATIOLI, Graciette; VALENTINI, Sóstenes Rosa; SOMMER, Willy Arno. Validação de métodos analíticos na quantificação de comprimidos de Captopril – comparação de metodologias para um programa de garantia de qualidade. Acta Scientiarum. Health Science, [s.l.], v. 26, n. 2, p.357-364, 4 abr. 2004. Universidade Estadual de Maringa. http://dx.doi.org/10.4025/actascihealthsci.v26i2.1590. Disponível em: <http://periodicos.uem.br/ojs/index.php/ActaSciHealthSci/article/view/1590>. Acesso em: 30 jul. 2017.
ROCHA, Tiago Galdino; GALENDE, Sharize Betoni. A importância do controle de qualidade na indústria farmacêutica. Revista UningÁ Review, Maringá, v. 20, n. 2, p.97-103, 13 out. 2014. Disponível em: <http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0100-40422009000800038>. Acesso em: 16 set. 2017.
SEVERINO, A. J. Metodologia do trabalho científico. São Paulo: Cortez, 2000. 279p.
SOUZA, Aline Mirelly Ferreira et al. Validação de limpeza de equipamentos multipropósito da linha de manipulação de comprimidos por granulação úmida: caso da furosemida. Revista de Ciências Farmacêuticas Básica Aplicada, Campina Grande, p.299-306, jan. 2012. Disponível em: <http://serv-bib.fcfar.unesp.br/seer/index.php/Cien_Farm/article/viewFile/1795/1247>. Acesso em: 08 mai. 2017.
TUBINO, M.; SIMONI, J. A. Refletindo sobre o caso Celobar®. Quím. Nova vol.30 no.2 São Paulo Mar./Apr. 2007.
VIEIRA, S.; HOSSNE, W. S. Metodologia científica para área de saúde. Rio de Janeiro: Elsevier, 2001. 192p.
VIEIRA, S. Com escrever uma tese. São Paulo: Pioneira, Thomson Learning, 2004. 102p.
[1] Discentes do curso de Farmácia da Uniararas – Fundação Hermínio Ometto
[2] Discentes do curso de Farmácia da Uniararas – Fundação Hermínio Ometto
[3] Docente e orientadora do curso de Farmácia da Uniararas – Fundação Hermínio Ometto
