ARTIGO DE REVISÃO
SALLES, Bruno Cesar Correa [1], OLIVEIRA, Carla Miguel de [2], MACIEL, Sarah Candido [3], DELMORO, Ana Carolina Lima [4], OLIVEIRA, Carlos Henrique Santos [5], CAMILO, Felipe Fidelis [6], TERRA, Michele Caroline [7]
SALLES, Bruno Cesar Correa. Et al. Uma Revisão De Literatura Sobre A Deficiência De Glicose-6-Fosfato-Desidrogenase. Revista Científica Multidisciplinar Núcleo do Conhecimento. Ano 06, Ed. 03, Vol. 13, pp. 91-105. Março de 2021. ISSN: 2448-0959, Link de acesso: https://www.nucleodoconhecimento.com.br/saude/literatura-sobre
RESUMO
A glicose-6-fosfato desidrogenase trata-se de uma enzima que está expressa em todas as células, sendo de extrema importância para a catalisação química da molécula nicotinamida adenina dinucleotídeo. Essa molécula atua na proteção, principalmente dos eritrócitos, contra agentes oxidantes, evitando a lise dessas células. Na deficiência da glicose-6-fosfato desidrogenase, a hemólise ocorre após febres, estresse oxidativo, infecção bacteriana ou viral aguda e cetoacidose diabética. Este trabalho tem como objetivo elaborar uma revisão bibliográfica, sobre a prevalência e as principais manifestações clínicas da deficiência da glicose-6-fosfato desidrogenase. Para isto, foram utilizados artigos publicados nos anos de 2000 a 2020, sendo originais, de língua portuguesa, espanhola e inglesa. O estudo trata-se de uma análise transversal, descritivo, que teve por base os seguintes bancos de dados: SciELO, PubMed, Mendely e Science Direct, utilizando os seguintes descritores: deficiência de glicose-6-fosfato desidrogenase, hemólise, radicais livres e nicotinamida adenina dinucleotídeo. Foram levantados 41 artigos científicos, sendo 4 na língua portuguesa, 35 na língua inglesa e 2 na língua espanhola. Do total de artigos levantados, 7,5% abordaram os conceitos bioquímicos da glicose-6-fosfato desidrogenase, 30% dos artigos publicados descrevem a deficiência da glicose-6-fosfato desidrogenase com portadores da malária, 27,5% abordaram a icterícia neonatal e anemia hemolítica, 15% sobre as variantes mutáveis e métodos laboratoriais com 15% dos artigos. A prevalência de deficiência foi abordada em 5% dos artigos levantados nesta revisão. De acordo com o levantamento bibliográfico realizado, a média de prevalência no Brasil foi de 2,5% no Sudeste, 4,75% no Sul e 9% no Norte. Essa proporção deve-se à diversidade de raças no Brasil onde a prevalência é em indivíduos afro-descendentes. Ainda, fica evidente que as principais manifestações clínicas levantadas pelos autores foram: quadros hemolíticos agudos e icterícia neonatal. Com base nos artigos analisados, apesar dos dados epidemiológicos não serem abrangentes, o conhecimento da deficiência de glicose-6-fosfato desidrogenase é um ponto forte a ser estudado, uma vez que poucos estudos traçam a prevalência da deficiência de glicose-6-fosfato-desidrogenase na população. Conhecer esse tema fornecerá dados epidemiológicos para uma melhor abordagem no prognóstico da deficiência analisando o risco-benefício de seu tratamento.
Palavras-chave: deficiência de glicose-6-fosfato desidrogenase, hemólise, radicais livres, nicotinamida adenina dinucleotídeo.
1. INTRODUÇÃO
De acordo com Cappellini e Fiorelli, (2008), a deficiência da enzima glicose-6-fosfato desidrogenase (G6PD) afeta mais de 400 milhões de pessoas no mundo. O gene que codifica a G6PD está localizado no braço longo do cromossomo X, um defeito genético devido a mutações ligadas ao gene, a sua caracterização bioquímica permite reconhecer mais de 140 mutações. Os sintomas clínicos aparentes na deficiência vão de icterícia neonatal a uma anemia hemolítica.
A G6PD é encontrada em todas as células do organismo, participando do primeiro passo da via de hexose monofosfato, oxidando a glicose-6-fosfato a 6-fosfogluconolactona, reduzindo o fosfato de dinucleótido de nicotinamida e adenina (NADP) a nicotinamida adenina dinucleotídeo (NADPH), que é de extrema importância contra o estresse oxidativo (CAPPELLINI; FIORELLI, 2008).
É predominante na África, Ásia, região do Mediterrâneo e Oriente Médio, sendo relatada também na América do Norte e do Sul e no norte da Europa, como citado pelos autores Cappellini e Fiorelli (2008). Técnicas desenvolvidas em 1931 são usadas até hoje, para descrever a atividade enzimática de G6PD. A OMS classificou essa atividade nas seguintes classes: classe I de 1%, classe II de 10%, classe III de 10-60% sendo considerada atividade normal, e classe V de 110% (STANTON et al., 2012).
Essa deficiência é uma das enzimopatias mais comum, conhecida como eritro-enzimática, quando afeta os eritrócitos. As eritroenzimopatias ocasiona em anemias hemolíticas com diferentes graus de gravidade, sendo a deficiência de glicose-6-fosfato desidrogenase (G6PD) a enzimopatia mais constante na via da pentose fosfato. Até o momento foram relatadas 160 mutações onde os defeitos encontram-se na estrutura e ou na função da enzima (MANZO et al., 2014).
De acordo com Luzzatto et al. (2016), a G6PD foi descoberta em torno do século XIX, onde médicos observaram quadros de anemia e hemoglobinúria em crianças que faziam a ingestão de feijão, surgindo o termo “favismo”, também chamado de anemia hemolítica. As variantes relatadas até agora somente 10% foram estudadas. O diagnóstico de G6PD resulta na observação da atividade enzimática e parâmetros hematológicos, sendo os casos mais graves em indivíduos de classe I e casos leves em indivíduos da classe V (ROSAS et al., 2020).
Desde o ano 1973, a prevalência da deficiência de G6PD mostra-se constante e de acordo com Beutler (2008) cerca de 300.000.000 de pessoas no mundo eram deficientes.
O conhecimento a respeito da deficiência da glicose-6-fosfato desidrogenase e suas complicações é importante para melhor taxa de sobrevida de indivíduos portadores da deficiência e a prevenção dos quadros hemolíticos. Portanto, o objetivo deste trabalho é avaliar a prevalência da deficiência de glicose-6-fosfato desidrogenase em diversos aspectos, de acordo com artigos científicos publicados em diferentes bancos de dados.
2. MATERIAL E MÉTODOS
O presente projeto de pesquisa não envolveu o uso de dados de seres humanos e não exigiu qualquer avaliação clínico-laboratorial, sendo assim, não foi necessário ser submetido ao Comitê de Ética. O trabalho tem como base uma revisão bibliográfica, desenvolvida através de artigos científicos. Os artigos analisados tiveram como critérios de inclusão, artigos publicados nos anos de 2000 a 2020, sendo originais, de língua portuguesa, espanhola e inglesa, abordando o tema: “Deficiência de glicose-6-fosfato desidrogenase”. O estudo trata-se de uma análise transversal, descritivo, que teve por base os seguintes bancos de dados: SciELO, PubMed, Mendely e Science Direct. A pesquisa no Banco de Dados teve como palavras-chave: deficiência de G6PD, hemólise, radicais livres e NADH.
Tabela 1: A tabela mostra um total de 40 artigos publicados entre 2000 a 2020 abordando os descritores de deficiência de G6PD, hemólise, radicais livres e NADH.

3. GLICOSE 6 FOSFATO DESIDROGENASE (G6PD)
3.1 ESTRUTURA E FUNÇÃO DA G6PD
O metabolismo eritrocitário é, em grande parte, dependente da enzima G6PD para obtenção de energia através da glicose convertida em açúcar, atuando também na redução da nicotinamida adenina dinucleotídeo (NADPH). Composta por 515 aminoácidos, a G6PD conta com aproximadamente 59 kDa de peso molecular, estando presente em todas as células do organismo, podendo variar sua concentração (CAPPELLINI; FIORELLI, 2008).
Segundo a OMS, em 1967 foram realizadas as primeiras recomendações para a determinação da deficiência de G6PD, mais de 400 variantes genéticas descritas por técnicas padronizadas, a maioria apresentando defeito enzimático. Esse defeito enzimático pode ocasionar uma mudança qualitativa devido a uma diminuição no número de moléculas enzimáticas (CAPPELLINI; FIORELLI, 2008).
Essa mudança qualitativa pode acarretar uma série de defeitos nas células, os eritrócitos mais do que outras células, são suscetíveis a essas consequências. No entanto, há uma explicação para isso. Quando os glóbulos vermelhos envelhecem perdem parte de suas funções, esse processo, na maioria das vezes, está relacionado com as mutações que comprometem a estabilidade da G6PD (LUZZATTO et al., 2016).
Pelo fato de não possuírem mitocôndrias, os glóbulos vermelhos necessitam da molécula NADPH para sua fonte de energia. Sabe-se então, que a produção de NADPH é de extrema importância para proteção dessas células contra agentes oxidantes, evitando a lise dos eritrócitos. Podendo também, servir de doador de elétrons, especialmente colesterol e ácidos graxos, e formação de óxido nítrico (MEHTA et al., 2000).
A via hexose monofosfato tem um papel importante nas células, onde inclui a produção de fosfatos pentoses para produção de compostos químicos como os nucleotídeos, a produção de açúcares de carbono e a redução do NADP+ a NADPH, que como já citado é de extrema importância para defesa da célula contra agentes oxidantes (RAMNANAN; STOREY, 2005). O NADPH responsável pela proteção da célula contra o estresse oxidativo está presente em três vias antioxidantes, sendo elas, a glutationa, tioredoxina e glutarredoxina (STANTON et al., 2012).
A primeira fase é caracterizada pela regeneração da glutationa (GSH) pela glutationa redutase (GR). A GSH é fundamental para diminuição do peróxido de hidrogênio e radicais livres nos glóbulos vermelhos. Essa redução da GSH realiza um papel de desintoxicação quando reage com peróxido de hidrogênio e peroxidases. Deste modo, a atividade da G6PD ajuda as células sanguíneas a suportarem o estresse oxidativo (MANZO et al., 2014).
Em um indivíduo com a deficiência de G6PD já instalada, não possui uma homeostase, ou seja, as células daquele organismo reduzem suas funções normais e os processos de desintoxicação não são mantidos. Com isso, as células ficam suscetíveis aos ataques dos radicais livres. O excesso desses radicais livres desencadeia diversos fatores como o envelhecimento e doenças relacionadas a idade (ULUSU, 2015).
3.2 GENÉTICA E BASE MOLECULAR
Estudos mostram que a incidência maior da deficiência de G6PD é em indivíduos do sexo masculino, devido o gene G6PD está localizado no braço longo do cromossomo X, com 13 éxons e 12 íntrons. Embora mulheres apresentem menos sinais clínicos frente a deficiência, algumas desenvolvem quadros de anemia hemolítica aguda grave (CAPPELLINI; FIORELLI, 2008).
Mais de 140 variantes mutáveis da G6PD já foram identificadas, com substituições de aminoácidos e base única. Em estudos com análise tridimensional foi possível observar, que as mutações ocorrem em torno dos exões 10 e 11, qualquer mutação que ocorre dentro ou ao redor desta propriedade resultam em variantes deficientes da G6PD. As variantes da G6PD determinadas até o momento foram descritas bioquimicamente através da atividade enzimática, padrões de mobilidade observando o comportamento pela cromatografia. (CAPPELLINI; FIORELLI, 2008).
De acordo com a OMS, essas variantes são reunidas em cinco classes: classe I com 1% de atividade enzimática ou nenhuma, tendo como as principais variantes G6PD Buenos Aires e G6PD Durham; classe II com 10% tendo como principal variante G6PD Mediterrâneo, classe III com 10-60% tendo como principal variante a G6PD A, classe IV apresentando 60-90% de atividade normal tendo como principal variante a G6PD Montalbano, e a classe V com 110% de aumento da atividade enzimática com nenhuma variante reportada (MINUCCI et al., 2009).
Essas variantes foram classificadas conforme com o grau de deficiência e suas manifestações clínicas, podendo variar de quadros mais graves com 5% de atividade residual na classe I e de forma mais branda na classe V. Mundialmente, 83,9% das variantes causam uma única substituição de aminoácidos. Sendo, 8,7% das variantes com mutações duplas e triplas identificadas com menor frequência, indicando que a deficiência da G6PD é uma anomalia heterogênea (ROSAS et al., 2020).
As regiões com maior prevalência da deficiência da G6PD possuem pelo menos algum tipo de variante mutável, como a variante G6PD A que apresenta cerca de 90% da deficiência, essa variante está presente em regiões com predominância de indivíduos de origem africana. A G6PD Mediterrâneo é a segunda variante mais comum de ser encontrada, sendo responsável pela maioria da deficiência nas populações ao redor do mar Mediterrâneo (CAPPELLINI; FIORELLI, 2008).
A variante G6PD A é a mais prevalente no Brasil, sendo caracterizada pela troca de nucleotídeos na posição 376, apresentando o polimorfismo A376G, também podendo ser chamada de variante G6PD africana. Os indivíduos que possuem essa variante mutável, possuem em média 48,71% de atividade enzimática normal, sendo um valor considerado para os padrões da variante G6PD A, que variam de 10 a 60% da atividade enzimática (PEREIRA et al., 2019).
3.3 EPIDEMIOLOGIA
A deficiência de G6PD é de longe a enzimopatia mais prevalente, atingindo mais de 400 milhões de pessoas em todo o mundo, em particular na África, sul da Europa, Oriente Médio, sudeste da Ásia e as ilhas do Pacífico central e do sul, afetando América do Norte e do Sul e algumas partes do norte da Europa. Estudos epidemiológicos mostraram-se imprecisos e não se ampliaram ao mundo todo, fazendo com que o prognóstico e tratamento seja ainda mais difícil (CAPPELLINI; FIORELLI, 2008).
A classificação das mutações varia de população, como as variantes G6PD A- e G6PD B- que afetam homens africanos e afro-americanos e populações do Mar Mediterrâneo respectivamente. Possui uma distribuição mundial de cerca de 4,9%, estima-se que a população mais afetada são indivíduos de ascendência africana, afetando até 10% da população do Mediterrâneo. Já na América Latina a deficiência de G6PD se expande nas regiões costeiras, mostrando uma prevalência menor do que na África e Ásia, sendo inferior a 1% (MANZO et al., 2014).
Suldrup et al (2014) revelam que a deficiência de G6PD é a anormalidade enzimática mais comum, sendo uma dificuldade para a saúde pública enfrentar. Estima-se que pelo menos 7,5% da população mundial possui um ou mais genes para essa anomalia. A proporção em diversos países, citada anteriormente, é devido às migrações que podem ocorrer em diversos países e regiões do mundo (KATSURAGAWA et al., 2004).
É considerado que a epidemiologia da deficiência de G6PD abrange o mundo todo. Alguns dados confirmam que a G6PD é a variante deficiente mais comum e que resulta em diversas combinações de mutações. Essas mutações reduzem a atividade da enzima G6PD, podendo agravar o quadro clínico do indivíduo (BEUTLER et al., 2007).
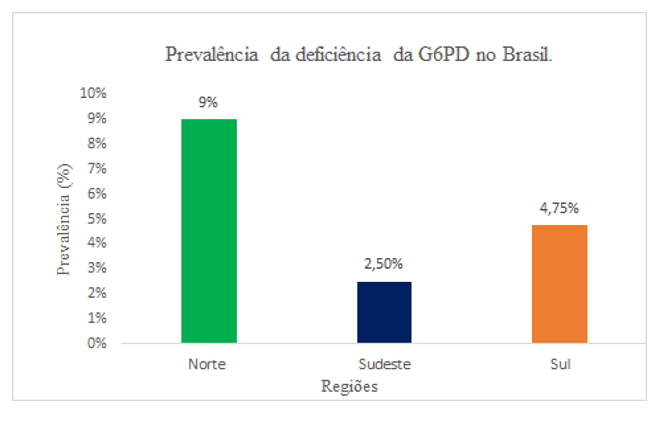
No Brasil, a deficiência da G6PD chega a afetar até 2,5% dos recém-nascidos, sendo um valor considerado para os padrões de estudo epidemiológicos da doença. Em três regiões do Brasil, foi observado uma prevalência de 2,5% no Sudeste, 4,75% no Sul e 9% no Norte. Outros estudos realizados obterão uma média de 3,59% de prevalência em adultos e crianças, sendo todos os indivíduos do sexo masculino identificados com a deficiência (PEREIRA et al., 2019) (Gráfico 1).
Gráfico 1: O gráfico representa a prevalência da deficiência de glicose-6-fosfato desidrogenase em três regiões do Brasil, em recém-nascidos no ano de 2019.
3.4 DIAGNÓSTICO E MANIFESTAÇÕES CLÍNICAS
Durante o século XX, diversos médicos no sul da Itália e Sardenha desvendaram o quadro clínico desencadeado pela ingestão de favas. Com isso, relacionaram o favismo como efeito tóxico a indivíduos deficientes de G6PD. De acordo com Cappellini e Fiorelli (2008), as hemólises que ocorrem em decorrência do favismo é consequência de uma série de substâncias tóxicas para pacientes com deficiência de G6PD, como a divicina, isouramil e convicina. O estresse oxidativo que ocorre nos pacientes após ingestão do feijão causa uma série de modificações nos eritrócitos, levando a um quadro de anemia hemolítica que se apresenta 24 horas após o consumo.
Assim como, alguns indivíduos podem se apresentar assintomáticos, outros desenvolvem quadros mais graves de estados hemolíticos agudos ou crônicos. A gravidade do quadro clínico na maioria das vezes está relacionada com o grau de deficiência enzimática. Um dos sinais clínicos em deficientes de glicose-6-fosfato desidrogenase é a icterícia neonatal, quadro clínico grave que afeta prematuros (MANZO et al., 2014).
O quadro mais comum é a hemólise aguda, ocorre entre 24 a 48 horas comumente após estresse oxidativo por ingestão de favas, drogas ou infecções. Estudos relataram também o desencadeamento dessa hemólise por diabetes, ataque cardíaco ao miocárdio e exercício físico intenso, podendo nenhum destes serem descartados (MANZO et al., 2014).
Existem diversos testes laboratoriais que são pressupostos no diagnóstico de G6PD, pautado na estimativa da atividade enzimática como a análise espectrofotométrica. Para um melhor diagnóstico da G6PD, diversos métodos semiquantitativos estão em desenvolvimento para determinação da atividade enzimática da G6PD. Estes incluem os métodos citados por Cappellini e Fiorelli (2008), teste quantitativo do formazan, reduzindo o tempo de preparação e simplificando a análise dos resultados (MANZO et al., 2014).
A OMS recomenda a descrição bioquímica da G6PD, através de métodos moleculares como a reação em cadeia da polimerase (PCR), sequenciamento direto, eletroforese em gel com gradiente desnaturante, ou seja, testes laboratoriais a fim de detectar mutações específicas na população. Algumas mutações ocasionam variantes que resultam em um quadro hemolítico severo ou até mesmo quadros assintomáticos (CAPPELLINI; FIORELLI, 2008).
A deficiência da G6PD se tornou um alerta para a OMS, devido à sua alteração metabólica heterogênea, onde seus portadores manifestam quadros de anemia hemolítica aguda induzida por agentes endógenos ou exógenos. De acordo com Compri et al. (2000), a deficiência de G6PD no Brasil não é um problema preferencial da saúde pública, apesar de possuir testes de triagem que são econômicos e eficientes, ainda dependem de uma demanda maior e orientações específicas para a população.
Os neonatos deficientes de G6PD, podem desenvolver quadros de icterícia neonatal, desse modo, os autores Iglessias et al. (2010) descrevem o teste de Brewer para recém-nascidos, onde ajuda a descrever a etiologia, permitindo uma intervenção antes da manifestação clínica. Com este teste de triagem é possível prever a icterícia neonatal e até mesmo identificar a deficiência de G6PD no seu nível de 6,0 UI, e impedir a prescrição de medicamentos oxidativos para os recém-nascidos.
A icterícia neonatal é evidente por volta de 1 a 4 dias após o nascimento do bebê, desenvolvendo um quadro de hiperbilirrubinemia. A hiperbilirrubinemia é caracterizada pelo aumento de bilirrubina no sangue, onde o fígado do recém-nascido ainda não consegue metabolizar o excesso dessa substância. Torna-se mais grave em recém-nascidos deficientes de G6PD, estudos apontam que a diminuição da conjugação de bilirrubina e a depuração hepática favorecem a hemólise (MANZO et al., 2014).
O aumento da bilirrubina no sangue do recém-nascido pode desencadear outras manifestações clínicas, como a Kernicterus, mesmo sendo rara de ocorrer, pode causar danos neurológicos permanentes. Na deficiência da G6PD, é constante o aparecimento de hemólise que resulta em um aumento de bilirrubina, os níveis elevados de bilirrubina podem desencadear lesões graves no sistema nervoso central (WATCHKO, 2017).
Mesmo em alguns casos assintomáticos, os portadores desta anormalidade genética poderão em algumas situações ambientais, apresentarem crises hemolíticas induzidas, que surgem quando as células sofrem estresse oxidativo provocados por agentes como drogas, infecção ou ingestão de favas. De acordo com Cappellini e Fiorelli (2008) a deficiência não afeta a expectativa e qualidade de vida.
Desde que a G6PD foi diagnosticada após o uso de primaquina, medicamento utilizado no tratamento da malária, diversos medicamentos provocaram o risco de hemólise em pacientes com a deficiência de G6PD, até mesmo a cloroquina, que também é utilizada no tratamento da malária, foi citada recentemente pelos autores (KUUPIEL et al., 2020).
Contudo, estudos apontam uma confusão entre o uso desses medicamentos em pessoas com uma infecção já preexistente, um dos principais motivos é que a própria infecção pode causar a hemólise, logo, muitos indivíduos foram submetidos a essas drogas já com o sinal clínico de uma anemia hemolítica desencadeada por uma infecção (LUZZATTO et al., 2016).
4. CONSIDERAÇÕES FINAIS
Foram levantados 41 artigos científicos sendo 4 na língua portuguesa, 35 na língua inglesa e 2 na língua espanhola. Do total de artigos levantados, 7,5% abordaram os conceitos bioquímicos da glicose-6-fosfato desidrogenase, 30% dos artigos publicados descrevem a deficiência da glicose-6-fosfato desidrogenase com portadores da malária, 27,5% abordaram a icterícia neonatal e anemia hemolítica, 15% de artigos as variantes mutáveis e métodos laboratoriais com 15% dos artigos. A prevalência de deficiência foi abordada em 5% dos artigos levantados nesta revisão. De acordo com nosso levantamento bibliográfico, observamos que a média de prevalência no Brasil foi de 2,5% no Sudeste, 4,75% no Sul e 9% no Norte. Essa proporção se deve a diversidade de raças no Brasil ocasionado pelas migrações, onde a prevalência maior é em indivíduos afrodescendentes.
REFERÊNCIAS
AINOON, O. et al. Glucose-6-Phosphate Dehydrogenase (G6PD) Variants in Malaysian Malays. HUMAN MUTATION Mutation in Brief, [S. l.], p. 1-9, 29 out. 2002.
AU, WY et al. Glucose-6-phosphate dehydrogenase deficiency and hematopoietic stem cell transplantation. Bone Marrow Transplantation, [S. l.], p. 399–402, 3 dez. 2002.
BALGIR, R. S. Community Expansion and Gene Geography of Sickle Cell Trait and G6PD Deficiency, and Natural Selection against Malaria: Experience from Tribal Land of India. Cardiovascular & Hematological Agents in Medicinal Chemistry, [S. l.], p. 3-13, 1 maio 2012.
BARISIÉ, Marin et al. Characterization of G6PD deficiency in southern Croatia: description of a new variant, G6PD Split. J Hum Gene, [S. l.], p. 547–549, 6 set. 2005.
BEUTLER, Ernest et al. Glucose-6-Phosphate Dehydrogenase Deficiency and Antimalarial Drug Development. The American Society of Tropical Medicine and Hygiene, [S. l.], p. 779-789, 1 maio 2007.
BEUTLER, Ernest. Glucose-6-phosphate dehydrogenase deficiency: a historical perspective. BLOOD, [S. l.], p. 16-23, 1 jan. 2008.
CAPOLUONGO, E et al. Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency. Brenner’s Encyclopedia of Genetics, [S. l.], p. 340-342, 1 abr. 2013.
CAPPELLINI, M D; FIORELLI, G. Glucose-6-phosphate dehydrogenase defi ciency. The lancet, [S. l.], p. 64-74, 5 jan. 2008.
CASTRO, Simone et al. Prevalence of G6PD deficiency in newborns in the south of Brazil. Journal of Medical Screening, [S. l.], p. 85–86, 2 mar. 2006.
COMPRI, Mariane B. et al. Programa comunitário e deficiência de G-6-PD no Brasil. Rev.bras.hematol.hemoter, [S. l.], p. 33-39, 1 maio 2000.
GALATAS, Beatriz et al. Heterogeneity of G6PD deficiency prevalence in Mozambique: a school-based cross-sectional survey in three different regions. Malaria Journal, [S. l.], p. 2-8, 1 maio 2017.
GOTSMAN, ISRAEL; MUSZKAT, MORDECHAI. Glucose-6-phosphate dehydrogenase deficiency is associated with increased initial clinical severity of acute viral hepatitis A. Journal of Gastroenterology and Hepatology, [S. l.], p. 1239–1243, 24 jul. 2001.
GUEYE, Nerly Shirère Gampio et al. An update on glucose-6-phosphate dehydrogenase defciency in children from Brazzaville, Republic of Congo. Malaria Journal, [S. l.], p. 1-6, 17 jun. 2019.
GUNAWARDENA, Sharmini et al. Prevalence of G6PD deficiency in selected populations from two previously high malaria endemic areas of Sri Lanka. PLOS ONE , [S. l.], p. 1-13, 2 fev. 2017.
HAMEL, Arno Rolf et al. Molecular Heterogeneity of G6PD Deficiency in an Amazonian Population and Description of Four New Variants. Blood Cells, Molecules, and Diseases, [S. l.], p. 399–406, 3 maio 2002.
HWANG , Sunhee et al. Correcting glucose-6-phosphate dehydrogenase deficiency with a small-molecule activator. NATURE COMMUNICATIONS, [S. l.], p. 1-12, 9 maio 2018.
IGLESSIAS, Marli Auxiliadora C. et al. Erythrocyte glucose-6-phosphate dehydrogenase deficiency in male newborn babies and its relationship with neonatal jaundice. REVISTA BRASILEIRA DE HEMATOLOGIA E HEMOTERAPIA, [S. l.], p. 434-438, 9 maio 2010.
JIANG, Peng et al. P53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. NATURE CELL BIOLOGY, [S. l.], p. 310-316, 11 fev. 2011.
KATSURAGAWA, Tony H.; GIL, Luiz H. Soares; STÁBILE, Rodrigo G.; PIRES, Matheus G.; DOMINGOS, Cláudia R. Bonini-. Avaliação da incidência da deficiência de Glicose-6-Fosfato Desidrogenase (G6PD) e perfil hematológico em indivíduos de uma região de Rondônia. Rev. bras. hematol. Hemoter. , [S. l.], p. 268-273, 4 abr. 2004.
KUUPIEL, Desmond et al. Geographical Accessibility to Glucose-6-Phosphate Dioxygenase Deficiency Point-of-Care Testing for Antenatal Care in Ghana. Diagnostics, [S. l.], p. 02-14, 16 abr. 2020.
LIU, Huajun et al. Association Between G6PD Deficiency and Hyperbilirubinemia in Neonates: A Meta-Analysis. Pediatric Hematology and Oncology, [S. l.], p. 92-98, 1 maio 2015.
LUZZATTO, Lucio et al. Favism and Glucose-6-Phosphate Dehydrogenase Deficiency. The new england journal o f medicine, [S. l.], p. 60-71, 4 jan. 2018.
LUZZATTO, Lucio et al. Glucose-6-Phosphate Dehydrogenase Deficiency. Hematol Oncol Clin, [S. l.], p. 374-393, 1 maio 2016
MANZO, Saúl Gómez- et al. Deficiencia de glucosa-6-fosfato deshidrogenasa: De lo clínico a lo bioquímico. Acta Bioquím Clín Latinoam, [S. l.], p. 410-420, 1 maio 2014.
MASON, Philip J; BAUTISTA, Jose´ M; GILSANZ, Florinda. G6PD deficiency: the genotype-phenotype association. Elsevier, [S. l.], p. 267–283, 2 maio 2007.
MEHTA, Atul et al. Glucose-6-phosphate dehydrogenase de®ciency. BaillieÁ re’s Clinical Haematology, [S. l.], p. 21-38, 1 maio 2000.
MINUCCI, Angelo et al. Glucose-6-phosphate Dehydrogenase Laboratory Assay: How, When, and Why?. IUBMB Life, [S. l.], p. 27-34, 5 jan. 2009.
MOCKENHAUPT, Frank P. et al. Reduced prevalence of Plasmodium falciparum infection and of concomitant anaemia in pregnant women with heterozygous G6PD deficiency. Tropical Medicine and International Health, [S. l.], p. 118–124, 26 fev. 2003.
NKHOMA, Ella T. et al. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: A systematic review and meta-analysis. Elsevier, [S. l.], p. 267-278, 23 fev. 2009.
PEREIRA, Lucas Luís Meigre Dias et al. Prevalência da deficiência de G6PD e caracterização molecular dos polimorfismos G202A, A376G e C563T em neonatos no Sudeste do Brasil. Einstein , [S. l.], p. 1-7, 24 jun. 2019.
RAMNANAN, Christopher J.; STOREY, Kenneth B. Glucose-6-phosphate dehydrogenase regulation during hypometabolism. Biochemical and Biophysical Research Communications, [S. l.], p. 7-16, 18 out. 2005.
ROSAS, Víctor Martínez- et al. Effects of Single and Double Mutants in Human Glucose-6-Phosphate Dehydrogenase Variants Present in the Mexican Population: Biochemical and Structural Analysis. International Journal Molecular Sciences, [S. l.], p. 02-23, 15 abr. 2020.
SANTOS, Sandna Larissa Freitas dos et al. Associação da deficiência da glicose-6-fosfato desidrogenase (G6PD) em população brasileira afrodescendente. Boletim Informativo Geum, [S. l.], p. 106-111, 17 mar. 2016.
SERPA, Jose A. et al. Prevalence of G6PD deficiency in a large cohort of HIV-infected patients. Elsevier, [S. l.], p. 399–402, 21 ago. 2010.
STANTON, Robert C. et al. Glucose-6-Phosphate Dehydrogenase, NADPH, and Cell Survival. IUBMB Lif, [S. l.], p. 362-369, 1 maio 2012.
SUKUMAR, Sridevi et al. Molecular basis of G6PD deficiency in India. Elsevier, [S. l.], p. 141 – 145, 30 jul. 2004.
SULDRUP, Niels Alejandro Federico et al. Deficiencia de glucosa-6-fosfato deshidrogenasa en recién nacidos en Argentina. Acta Bioquím Clín Latinoam, [S. l.], p. 170-182, 1 maio 2014.
TANTULAR, Indah S.; KAWAMOTO, Fumihiko. An improved, simple screening method for detection of glucose-6-phosphate dehydrogenase deficiency. Tropical Medicine and International Health, [S. l.], p. 569-574, 18 jun. 2003.
ULUSU, N. Nuray. Glucose-6-phosphate dehydrogenase deficiency and Alzheimer’s disease: Partners in crime? The hypothesis. Elsevier, [S. l.], p. 219-223, 5 maio 2015.
WATCHKO, Jon F. Refractory Causes of Kernicterus in Developed Countries: Can We Eradicate G6PD Deficiency Triggered and Low-Bilirubin Kernicterus?. Current Pediatric Reviews, [S. l.], p. 159-168, 7 jul. 2017.
ZHU, Aiping et al. An enzymatic colorimetric assay for glucose-6-phosphate. Elsevier, [S. l.], p. 266-270, 27 ago. 2011.
[1] Doutor em Ciências Farmacêuticas.
[2] Mestrado em Ciências Farmacêuticas.
[3] Graduanda de Biomedicina.
[4] Graduanda de Farmácia.
[5] Graduando de Farmácia.
[6] Graduando de Farmácia.
[7] Graduanda de Biomedicina.
Enviado: Janeiro, 2021.
Aprovado: Março, 2021.

Uma resposta
Olá! Gostaria muito de saber se posso obter de alguma forma informações mais detalhadas sobre a mutação do meu filho já fiz teste genético e ele tem 2 mutação no x estou preocupada pq ele pode ter a doença mais grave nos somos do Rio de Janeiro e se possível como podemos encontrar o autor do artigo para uma conversa?! Preciso de um respaldo científico para tratar meu filho. Obrigada