ARTIGO ORIGINAL
LIMA, Tallison Renan Tenorio [1], AMORIM, Thiago Ottoni Wanderley [2], CRUZ, José Anderson da Silva [3]
LIMA, Tallison Renan Tenorio. AMORIM, Thiago Ottoni Wanderley. CRUZ, José Anderson da Silva. Feocromocitoma assintomático em uma paciente jovem com incidentaloma adrenal. Revista Científica Multidisciplinar Núcleo do Conhecimento. Ano 03, Ed. 11, Vol. 01, pp. 48-56 Novembro de 2018. ISSN:2448-0959
RESUMO
Feocromocitomas são tumores de células cromafins da medula adrenal que produzem, armazenam, metabolizam e secretam catecolaminas. Podem ocorrer em qualquer idade, com um pico de incidência entre a quarta e a quinta década, sendo raros após os 60 anos. Aproximadamente 10% dos feocromocitomas são diagnosticados como massas adrenais clinicamente inaparentes. Sua apresentação clínica é bastante variável, incluindo sinais e sintomas relacionados com o excesso de catecolaminas e sua tríade clássica consiste em hipertensão grave, cefaleia intensa e palpitações. São infrequentes em indivíduos assintomáticos. O presente relato descreve o caso de uma paciente jovem assintomática que, após a realização exames de imagem, evidenciou massa de grandes dimensões em suprarrenal. Exames laboratoriais e de imagens confirmaram o diagnóstico. A paciente, após preparo adequado, foi submetida à ressecção cirúrgica do feocromocitoma. Este caso ilustra a importância de se pesquisar feocromocitoma em qualquer paciente portador de incidentaloma adrenal.
Palavras-chave: Feocromocitoma, incidentaloma adrenal, paciente jovem.
INTRODUÇÃO
Os feocromocitomas são tumores produtores de catecolaminas, preferencialmente adrenalina e/ou noradrenalina (NA), originados das células cromafins do eixo simpático-adrenomedular. Em decorrência da produção endócrina, o tumor pode desencadear grave hipertensão arterial sustentada ou paroxística, devendo ser considerado sempre em pacientes com hipertensão arterial intermitente e resistente ao tratamento anti-hipertensivo convencional (Faiçal e Shiota, 1997; Vinícius e Malachias, 2002). Além da hipertensão, comumente paroxística, portadores do tumor tem como manifestações clínicas palpitações, sudorese e cefaléia, resultantes da hiperestimulação simpática (Darzé, Sohsten e Atlantis, 2004). Entretanto, em casos mais raros, os pacientes com feocromocitoma podem ser assintomáticos, sendo diagnosticados acidentalmente após realização de exames de imagens por outras razões (Bovio et al., 2006). Reportamos aqui o caso de uma paciente jovem assintomática que, após realização de exames de imagem, evidenciou feocromocitoma como achado incidental.
MÉTODO
Trata-se de um estudo descritivo do tipo Relato de Caso, utilizando-se de informações obtidas durante assistência médica em uma unidade de endocrinologia de um hospital de referência do estado de Alagoas.
DESCRIÇÃO DO CASO
Paciente do sexo feminino, 29 anos, foi encaminhada ao ambulatório de endocrinologia após achado tomográfico de massa em topografia adrenal direita. Apresentou-se sem queixas no momento da consulta e negando história de hipertensão arterial sistêmica. Foram solicitados exames complementares laboratoriais que apresentaram os seguintes resultados: Adrenalina 11,3 mcg/24h; Noradrenalina 168,3 mcg/24h; Normetanefrina em urina de 24 horas > 4.260 mcg/L; Metanefrina em urina de 24 horas 216,3mcg; Cortisol sérico após supressão (1mg de dexametasona às 23 horas) 1,06 mcg/dL. Além da solicitação de exames de imagem: Ressonância magnética (Imagem 1) de abdômen superior que apresentou os achados: presença de formação nodular, predominantemente hipointensa em T1 e hiperintensa em T2, de contornos regulares, com realce heterogêneo na topografia da glândula adrenal direita após contraste venoso de gadolínio, medindo 5,5 x 5,4 x 5,3 cm; Cintilografia de corpo inteiro com MIBG 131 (Imagem 2) (metaiodobenzilguanidina) que evidenciou hipercaptação do radiofármaco em região da glândula adrenal direita. Com os resultados laboratoriais e por exames de imagem foi confirmado o diagnóstico de feocromocitoma, seguido da investigação para Neoplasias endócrinas múltiplas (NEM) 2A e 2B (mandatório quando confirmado o diagnóstico de feocromocitoma), porém os achados clínicos e laboratoriais descartaram a presença de NEM 2A ou 2B. Após preparo, a paciente foi submetida a cirurgia para ressecção do tumor.
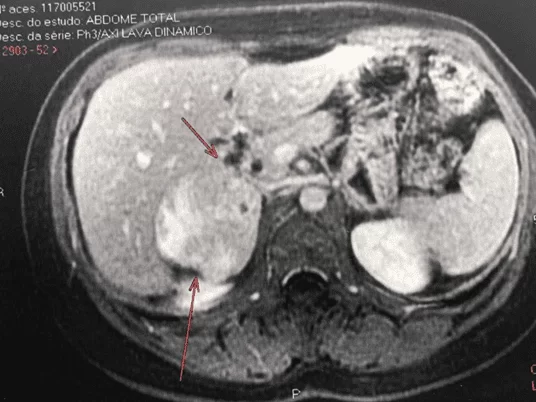
Imagem 1: Ressonância magnética do abdômen ponderada em T2 demonstrando formação nodular hiperintensa, de contornos regulares, com realce heterogêneo na topografia da glândula adrenal direita após contraste venoso de gadolínio, medindo 5,5 x 5,4 x 5,3 cm.
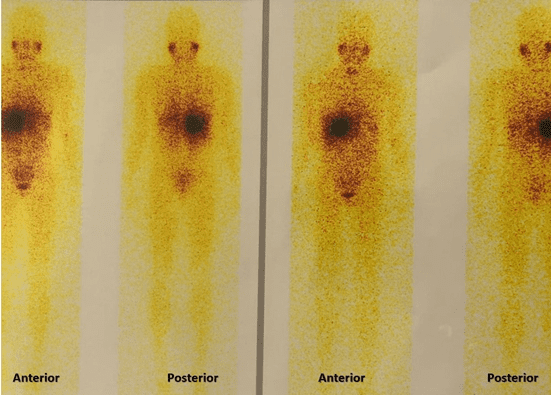
Imagem 2: Cintilografia de corpo inteiro com MIBG 131 evidenciando hipercaptação do radiofármaco em região da glândula adrenal direita.
DISCUSSÃO
O feocromocitoma é um tumor com estimativa de incidência anual de 2 a 8 casos por 1 milhão de habitantes, acomete ambos os sexos em igual proporção, surgindo comumente entre a 4ª e 5ª década de vida, sendo raro em pessoas da raça negra. Em 80 a 90% dos casos correspondem a neoplasias benignas, comumente unilateral, sendo bilateral em apenas 3 a 11% dos casos, a incidência bilateral é muito mais expressiva nos tumores de origem familial (Costa e Gomes, 2008). A maioria dos feocromocitomas localiza-se na adrenal (90% dos casos), sendo os demais extra-adrenais chamados comumente de paragangliomas, que podem ter origem em paragânglios da base do crânio até a bifurcação das artérias ilíacas, órgão de Zuckerkandl, mais raramente em tórax, bexiga ou cérebro. A frequência de malignidade dessas neoplasias é de 10%, porém em localizações fora das adrenais pode ocorrer em 20% a 40% dos casos, quando malignos, estes tumores apresentam metástases frequentemente em ossos, linfonodos regionais, fígado, pulmões, cérebro e cordão espinhal (Faiçal e Shiota, 1997; Vinícius e Malachias, 2002).
Embora a maioria dos casos de feocromocitoma seja esporádica, como a do caso apresentado, aproximadamente 10 – 20% são de origem hereditária. A maior parcela dos esporádicos são encontrados na topografia das glândulas adrenais, seguidos do abdômen e pélvis. Existem correlações já conhecidas de alterações nas sub-regiões cromossômicas 1p, 3p, 17p e 22q em pacientes portadores de feocromocitomas. Estudos identificaram três áreas de deleção no cromossomo 1p34-36, denominadas PC1, PC2 e PC3, sugerindo que possivelmente três genes supressores de tumor distintos estão relacionados à patogênese dos feocromocitomas esporádicos (Benn et al., 2001).
Pelo menos 20% dos casos de feocromocitoma podem ser causados por mutações em linhagens germinativas (feocromocitoma familial), devendo o diagnóstico seguir de acordo com as características fenotípicas do paciente em questão (Pires, 2017). Os pacientes podem apresentar feocromocitoma, câncer medular da tireoide (CMT) ou hiperplasia de células C e, em 50% dos casos, associados a hiperplasia ou neoplasia de paratireóide tem o que se chama de neoplasias endócrinas múltiplas (NEM) do tipo 2a. Já os portadores de feocromocitoma, CMT e ganglioneuromas do trato gastrointestinal possuem o NEM do tipo 2b. Além disso, doença de Von Hippel-Lindau (VHL), neurofibromatose tipo 1 e tumores do corpo carotídeo são associados a feocromocitoma familial. O tumor de células cromafins coexiste com hemangioblastomas do sistema nervoso central e angioma da retina em 14% dos pacientes. Portanto, deve ser pesquisado feocromocitoma quando alguma das condições citadas estiver presente e o contrário também se aplica. Quando for estabelecido a presença de feocromocitoma familial, em parentes de primeiro grau deve ser pesquisado a presença mutações genéticas como receptor de tirosina quinase de proto-oncogne (RET) no cromossomo 10 para NEM tipo 2a ou 2b, no cromossomo 3p para VHL, no cromossomo 17q para neurofibromatose familial e no cromossomo 11q para tumores de corpos carotídeos. Além disso, mutações na succinato desidrogenase foram identificadas em feocromocitomas aparentemente esporádicos (Manger, 2006). Portadores de mutações citadas devem ser acompanhados periodicamente.
A grande variabilidade de apresentação dos sintomas, pouco específicos, associado à baixa prevalência da patologia torna o diagnóstico difícil. A clínica é muito variável, de manifestações severas e dramáticas em alguns casos raros, como o reportado, a mínimas ou sem qualquer sintomatologia em outros, elevando de forma dramática a mortalidade. As manifestações clínicas, quando presentes, são relacionadas com a liberação pontual de catecolaminas na circulação, variando, desta forma, proporcionalmente à quantidade de catecolaminas (Li et al., 2015).
Diferentes tipos tumorais secretam níveis diferentes de cada catecolamina, apresentando quadros clínicos diferentes entre si. A tríade clássica consiste em cefaleia, diaforese e palpitações (Li et al., 2015; Pires, 2017). A hipertensão arterial é uma das manifestações mais frequentes, que levam o paciente a procura de serviço médico, e pode ser paroxística ou sustentada, sendo esta proporcionalmente inferior a primeira. Nos pacientes com tumores secretores predominantemente de epinefrina ou dopamina, sintomas de hipotensão ortostática, como pré-síncope, síncope e vertigens, são mais prevalentes. eitos da HTA e da vasoconstrição coronária podem resultar em cardiomiopatia ou mesmo numa forma de miocardite tóxica. Choque cardiogênico, edema agudo do pulmão, arritmias cardíacas e cardiomiopatia são vistos especialmente em doentes com tumores de grandes dimensões. Todas as repercussões são geralmente reversíveis após a cura cirúrgica (Kitayama et al., 2015; Li et al., 2015; Pires, 2017).
As catecolaminas são produzidas normalmente pelo sistema nervoso simpático e pela medula adrenal, não sendo, portanto, específicos do feocromocitoma (Sánchez Turcios, 2010). Em indivíduos normais, a epinefrina representa 80 a 85% das catecolaminas produzidas pela medula adrenal, o restante corresponde a noraepinefrina. A adrenal produz quantidades maiores de noradrenalina apenas em situações de estresse intenso. Por outro lado, em indivíduos com tumor de células cromafins, a produção de noradrenalina é, geralmente, superior a adrenalina. O tumor secreta as catecolaminas, comumente, episodicamente e, por isto, os níveis plasmáticos e urinários das catecolaminas são, geralmente, normais entre os episódios. Além disso, em cerca de 5 a 15% dos pacientes com feocromocitoma os níveis séricos e urinários de noradrenalina e adrenalina são normais. Após sua liberação/ação as catecolaminas são degradadas por ação enzimática da catecol-O-metil transferase (COMT) e monoamina oxidase (MAO) em seus metabólitos inativos chamados de metanefrinas (metanefrinas e normetanefrinas) e ácido vanilmandélico (VMA) (Sánchez Turcios, 2010).
A suspeita clínica de feocromocitoma é facilmente cogitada quando o paciente apresenta o quadro clínico característico, mas pode não ser cogitada nos casos atípicos ou assintomáticos e o tumor pode não ser detectado, com consequências por vezes letais para o paciente. Em assintomáticos, como o caso reportado, o diagnóstico é dificultado pela ausência de sintomas característicos, podendo provocar uma série de morbidades, evoluindo até para morte súbita por um pico hipertensivo. Com o intuito de evitar isso, é fundamental a investigação para exclusão ou confirmação da existência do tumor, mesmo que seja improvável. No diagnóstico diferencial de feocromocitoma é essencial considerar várias condições clínicas como hipertensão arterial essencial, ansiedade, síndrome do pânico, hiperplasia adrenomedular primária, hipertireoidismo, taquicardia paroxística, menopausa, enxaqueca, lesão intracraniana, epilepsia diencefálica, eclâmpsia ou pré-eclâmpsia, hipertensão por inibidores da MAO, hipoglicemia, neuroblastoma, ganglioneuroblastoma, infecções agudas, falência barorreflexa, ingestão de drogas (anfetamina, cocaína, ácido lisérgico, efedrina, fenilpropanolamina), suspensão de clonidina, insônia familiar fatal entre outras. Apesar da maioria dessas doenças ser facilmente identificada ou descartada pela história clínica e exames laboratoriais corriqueiros, algumas vezes é necessário o diagnóstico diferencial com feocromocitoma.
O diagnóstico é baseado na demonstração de produção autônoma de catecolaminas e/ou seus metabólitos, que pode ser feito pela dosagem de adrenalina e noradrenalina séricas e urinárias, metanefrinas e normetanefrinas (genericamente chamadas de metanefrinas) plasmáticas e urinárias e ácido vanilmandélico (VMA) urinário, além de testes funcionais e métodos de imagem (Faiçal e Shiota, 1997; Vinícius e Malachias, 2002). Os níveis de catecolaminas e metanefrinas no plasma e urina e VMA urinário sofrem influência de substâncias e fatores estressores como stress e doenças, por isto a dosagem isolada de um deles pode não ser suficiente para o diagnóstico de feocromocitoma, devendo feita a dosagem de mais de uma delas repetido pelo menos duas vezes associada a outros exames como testes funcionais e de imagem (Faiçal e Shiota, 1997; Peaston e Lai, 1993; Sánchez Turcios, 2010). Os níveis plasmáticos e urinários de metanefrina e normetanefrina são os testes mais sensíveis para diagnóstico tanto nos casos esporádicos quanto nos de origem familial (Manger, 2006), porém a dosagem das metanefrinas e normetanefrinas plasmáticas não é feita rotineiramente no Brasil, apenas a urinárias (Faiçal e Shiota, 1997; Vinícius e Malachias, 2002).
As recomendações atuais para o diagnóstico bioquímico feocromocitoma baseiam-se nas determinações de metanefrinas no sangue ou urina de 24 horas, de acordo com a disponibilidade. Valores acima de 3 a 4 limite superior de normalidade confirmam o diagnóstico de tumor de células cromafins. Valores normais desses compostos excluem o diagnóstico na vigência de hipertensão ou paroxismos, mas não pode ser excluído na ausência de sinais adrenérgicos quando dosados. Valores intermediários, entre o limite superior e 3 a 4 vezes acima dele, estão na “zona cinzenta” e podem representar resultados falso-positivos, pois concentrações elevadas desse hormônio não indicam, necessariamente, a presença do tumor e podem expressar apenas atividade aumentada do sistema nervoso autônomo simpático. Nesses casos os testes funcionais podem ser úteis. O uso de testes funcionais é importante quando a clínica é sugestiva e os níveis de catecolaminas e seus metabólitos não são elucidativos no diagnóstico de feocromocitoma. Um dos testes utilizados é o teste de supressão com clonidina (0,3 mg por via oral), já que a administração da clonidina reduz os níveis plasmáticos de catecolaminas e seus metabólitos no plasma em até três horas nos pacientes com hipertensão essencial, porém isso não ocorre nos feocromocitomas que permanecem elevados. Redução em 50% ou a níveis normais de noradrenalina após a supressão ocorre em 67% dos pacientes que não tem feocromocitoma, quando ocorre a redução dos níveis plasmáticos de normetanefrinas em mais de 40% ou a níveis normais exclui o diagnóstico de feocromocitoma em 96% dos casos, sendo um teste muito importante para exclusão de tumor de células cromafins (Faiçal e Shiota, 1997; Manger, 2006).
Além disso, outro teste funcional pode ser utilizado para pacientes em que se existe um forte indício da presença de feocromocitoma, na ausência de níveis elevados de catecolaminas e seus metabólitos, que é o teste de estímulo com glucagon. Esse teste não é muito usado, pois pode induzir uma crise hipertensiva grave e suas consequências, porém apresenta uma sensibilidade de 100% e especificidade de 81%, exceto em casos malignos e familiais em que perde em sensibilidade. A estimulação com glucagon em pessoas hígidas não é capaz de causar a liberação de catecolaminas, porém no feocromocitoma causa um aumento nos níveis de catecolaminas em mais de três vezes ou acima de 2.000 pg/mL em até três minutos (Faiçal e Shiota, 1997; Manger, 2006; Vinícius e Malachias, 2002). Após o diagnóstico laboratorial de feocromocitoma, o tumor deve ser localizado por exames de imagem (Manger, 2006).
Os exames de imagem devem ser direcionados, inicialmente, na topografia das glândulas adrenais, pois a maioria dos tumores são aí encontrados. Como primeira escolha surge a TC, podendo também a RMN ser usada, que permitem avaliar a localização, a ressecabilidade e evidenciar metástases locais. Na TC, os feocromocitomas podem ter grandes variações. Achados característicos incluem uma massa sólida de aspeto tipicamente heterogêneo, com necrose e calcificação comumente presentes. A TC contrastada ajuda na distinção de outras massas adrenais, onde se verifica intensa captação na fase arterial (>110 UH) e atraso no washout (<50% aos 10 minutos). Para tumores extra-adrenais, a RMN é uma melhor escolha. Não necessita de radiação ionizante ou contraste IV e é preferida em grávidas e crianças. Contudo, é mais dispendiosa e demorada, com uma resolução inferior. Os exames de imagem funcional – cintilografias (MIBG e SRS) e a PET (18F-FDG ou 18F-FDA) – constituem outras modalidades disponíveis. A MIBG tem uma estrutura semelhante à norepinefina e é captado especificamente pelo tecido adrenérgico (Manger, 2006; Pires, 2017; Vinícius e Malachias, 2002).
O tratamento de todos os feocromocitomas é cirúrgico e é altamente necessário em todos os tumores funcionais. Deve-se, entretanto, manter tratamento clínico farmacológico dos efeitos causados pelas catecolaminas circulantes do momento diagnóstico até o tratamento cirúrgico. Para redução de complicações cirúrgicas e pós-cirúrgicas, opta-se pelo bloqueio adrenérgico, realizando no mínimo duas semanas antes da cirurgia, e expansão do volume intravascular (Pires, 2017). O tratamento cirúrgico é baseado na adrenalectomia, e é o procedimento curativo padrão dos feocromocitomas. Em casos de ausência de síndrome hereditária, baixo risco de bilateralidade e tumores grandes, a adrenalectomia total é a indicação. Contudo, quando estão associados síndromes genéticos ou o risco de bilateralidade é elevado, a adrenalectomia parcial que poupa o córtex é uma alternativa (em especial NEM2 e VHL, cujo potencial metastático é baixo), eliminando a necessidade de terapia de substituição vitalícia. A via cirúrgica pode ser aberta ou laparoscópica, a depender do tamanho do tumor e da habilidade do cirurgião (Iacobone et al., 2017).
CONCLUSÃO
Feocromocitomas são tumores raros e de elevada mortalidade, sendo malignos em cerca de 10% dos casos. Deve-se pensar nesta neoplasia em pacientes com hipertensão de início recente, principalmente em jovens com sintomas de excesso de catecolaminas e pacientes com história familiar sugestivo de causa genética.
A paciente exposta no caso descrito apresentava-se totalmente assintomática, sendo o feocromocitoma um achado incidental de exame de imagem realizado. Entretanto, na investigação, possuía índices de catecolaminas séricas e urinárias acima do normal e hipercaptação do MIGB na topografia da glândula adrenal, fomentando o avanço para o diagnóstico. O tratamento baseou-se na adrenalectomia radical, com o restabelecimento dos níveis normais de catecolaminas circulantes e urinárias.
Feocromocitomas assintomáticos, achados como incidentaloma de adrenal, são raros e associados a morbidade e mortalidade consideráveis, devendo suspeitar desta neoplasia mesmo em pacientes assintomáticos, buscando a correção cirúrgica o mais breve possível.
REFERÊNCIAS
BENN, D.; DWIGHT, T.; RICHARDSON, A.; DELBRIDGE, L.; BAMBACH, C.; STOWASSER, M.; GORDON, R.; MARSH, D.; ROBINSON, B. Sporadic and familial pheochromocytomas are associated with loss of at least two. Mol Cell Endocrinol, v. 172, n. 1–2, p. 223–233, 2001.
BOVIO, S. et al. Prevalence of adrenal incidentaloma in a contemporary computerized tomography series. Journal of Endocrinological Investigation, v. 29, n. 4, p. 298–302, 2006.
COSTA, L.; GOMES, A. T. Feocromocitoma. Arquivos de Medicina, v. 22, n. 6, p. 177–187, 2008.
DARZÉ, E. S.; SOHSTEN, R. L. VON; ATLANTIS, B. A. Crise de Feocromocitoma Simulando um Infarto Agudo do Miocárdio em Paciente com Artérias Coronárias Normais. v. 82, n. no 2, p. 175–177, 2004.
FAIÇAL, S.; SHIOTA, D. Feocromocitoma: atualização diagnóstica e terapêutica. Revista da Associação Médica Brasileira, v. 43, n. 3, p. 237–244, 1997.
IACOBONE, M.; CITTON, M.; VIEL, G.; SCHIAVONE, D.; TORRESAN, F. Surgical approaches in hereditary endocrine tumors. Updates in Surgery, v. 69, n. 2, p. 181–191, 2017.
KITAYAMA, K. et al. A case of bilateral pheochromocytoma during pregnancy. BMC Surgery, v. 15, n. 1, p. 1–5, 2015.
LI, S.; LI, H.; JI, Z.; YAN, W.; ZHANG, Y. Primary adrenal teratoma: Clinical characteristics and retroperitoneal laparoscopic resection in five adults. Oncology Letters, v. 10, n. 5, p. 2865–2870, 2015.
MANGER, W. M. Diagnosis and management of pheochromocytoma – Recent advances and current concepts. Kidney International, v. 70, n. SUPPL. 104, 2006.
PEASTON, R. T.; LAI, L. C. Biochemical detection of phaeochromocytoma: Should we still be measuring urinary HMMA? Journal of Clinical Pathology, v. 46, n. 8, p. 734–737, 1993.
PIRES, M. Feocromocitoma : do espetro clínico ao tratamento. 2017.
SÁNCHEZ TURCIOS, R. A. Feocromocitoma: Diagnóstico y tratamiento. Revista Mexicana de Cardiología, v. 21, n. 3, p. 124–137, 2010.
VINÍCIUS, M.; MALACHIAS, B. Feocromocitoma – diagnóstico e tratamento. v. 9, n. 31, p. 160–164, 2002.
[1] Acadêmico da Faculdade de Medicina da Universidade Federal de Alagoas. Universidade Federal de Alagoas.
[2] Acadêmico da Faculdade de Medicina da Universidade Federal de Alagoas
[3] Titular em Endocrinologia e Metabologia pela Sociedade Brasileira de Endocrinologia e Metabologia. Professor da Faculdade de Medicina da Universidade Federal de Alagoas.
Enviado: Outubro, 2018
Aprovado: Novembro, 2018
