SICCHAR, Ana Cristina Suarez [1], GOMES, Marilu Cavalcante [2], LUSTOSA, Jéssica Ferreira [3], BATISTA, Aline Nogueira[4], RABELO, Ronaldo Marques Pontes [5]
SICCHAR, Ana Cristina Suarez. Et al. Morte súbita de origem cardíaca (por síndrome de Brugada) abortada: relato de caso. Revista Científica Multidisciplinar Núcleo do Conhecimento. Ano 03, Ed. 09, Vol. 1, pp. 123-130, Setembro de 2018. ISSN:2448-0959
INTRODUÇÃO
A morte súbita de origem cardíaca, definida como a cessação inesperada dos batimentos cardíacos com colapso hemodinâmico, é uma situação relativamente comum, mas desesperadora para os familiares e a equipe de assistência. Em geral ela ocorre devido surgimento de taquicardia ventricular sustentada ou fibrilação ventricular, no contexto de uma doença cardíaca estrutural, coronariopatia (causas mais comuns), ou, mais raramente, devido doenças genéticas2. Neste relato, é apresentado o caso de um paciente jovem, que apresentou morte súbita de origem cardíaca abortada, cuja investigação etiológica encontrou uma síndrome incomum, a síndrome de Brugada (SB).
DESENVOLVIMENTO DO RELATO DE CASO
Paciente do sexo masculino, 22 anos de idade, apresentou parada cardiorrespiratória sem causa óbvia, sem relação com esforço físico, em Manacapuru/AM; foi levado ao pronto-socorro local e recebeu medidas de suporte iniciais. Após retorno à circulação espontânea, evoluiu com choque de padrão cardiogênico, rebaixamento do nível de consciência e crises epilépticas recorrentes, e assim foi transferido para unidade de terapia intensiva (UTI), em Manaus/AM. Na história pregressa, havia relato de transtorno de personalidade, uso de cocaína e etilismo abusivo, e episódios de dor torácica sem investigação etiológica.
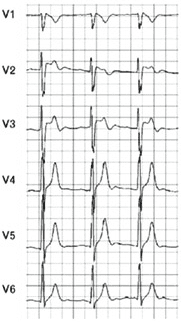
A internação na UTI durou 10 dias; o quadro neurológico e a disfunção hemodinâmica foram revertidos, ocorreram hepatite isquêmica e disfunção renal auto-limitados, sem outras disfunções orgânicas. O paciente foi, então, transferido para serviço especializado em cardiologia. Durante a investigação, eletrocardiogramas seriados não mostraram alterações de frequência, eixo, ritmo ou dos intervalos, mas a morfologia do complexo QRS nas derivações V1-2 possuía padrão rSr’, também chamado de pseudo-bloqueio de ramo direito ou “em sela”, e havia alteração transitória do segmento ST de V1-4 (vide figuras 1 e 2), sugestivos de síndrome de Brugada. Ecocardiograma transtorácico mostrou fração de ejeção de ventrículo esquerdo de 72%, sem disfunção sistólica segmentar nem dilatação ou hipocinesia de ventrículo direito. Estudo eletrofisiológico confirmou a hipótese diagnóstica, e um cardiodesfibrilador implantável (CDI) foi instalado no mesmo tempo operatório. O paciente recebeu alta para domicílio e permanece assintomático durante seguimento ambulatorial.
Figura 1. ECG do paciente relatado, mostrando o pseudo-BRD, supradesnível do segmento ST em V2, e padrão tipo 1 do segmento ST.
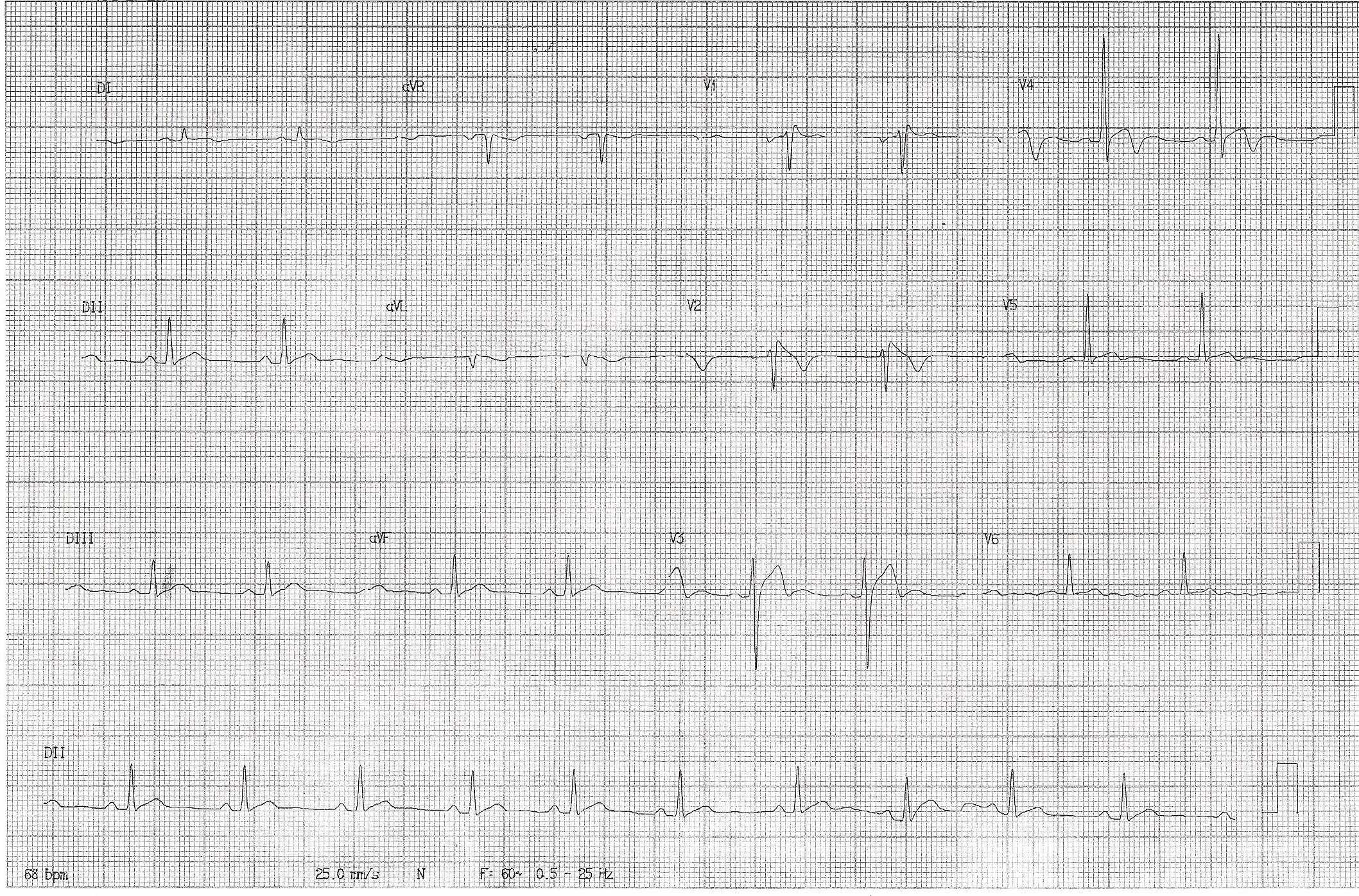
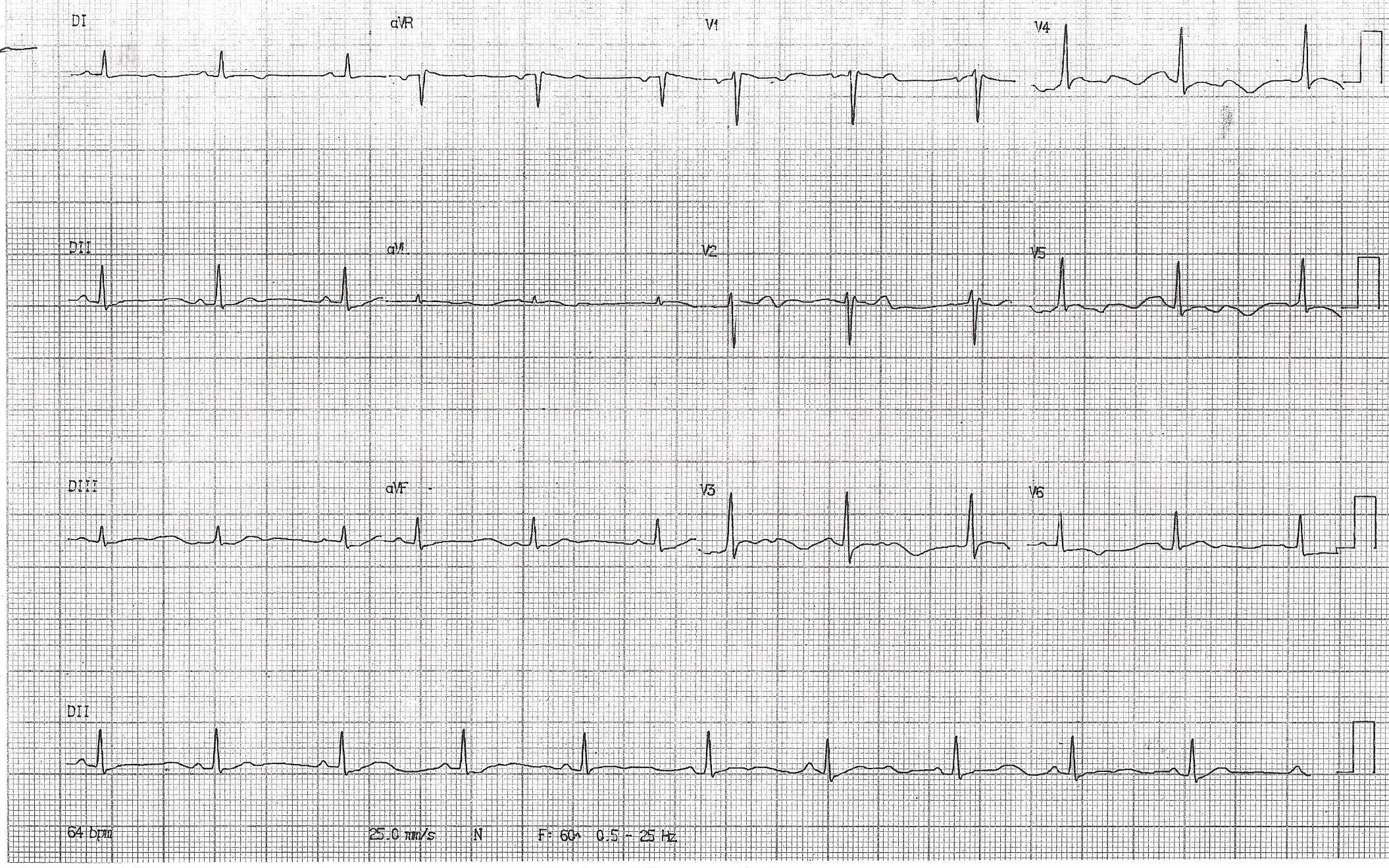
Figura 2. ECG do mesmo paciente em outro momento, mostrando o padrão tipo 2 do segmento ST (vide discussão).
DISCUSSÃO DO CASO
A SB é uma síndrome genética, de genótipo e fenótipo variáveis, de herança autossômica dominante e expressividade variável, cujo quadro clínico se compõe de eletrocardiograma com alterações típicas mais episódios de taquiarritmias atriais e ventriculares, síncope, respiração agônica noturna ou morte súbita de origem cardíaca3. Chamam-se portadores do padrão de Brugada (pB) aqueles pacientes assintomáticos que apresentam eletrocardiograma sugestivo4. A prevalência ao redor do mundo de pB é maior no Japão, ainda que inferior a 1% da população5. O risco de desenvolver eventos cardíacos nestes pacientes é de aproximadamente 10% a cada 2,5 anos6. O sexo masculino é mais afetado7; o diagnóstico é dado usualmente na idade adulta8.
A fisiopatogenia é multifatorial: perda de função em subunidades de canais de sódio dos miócitos, por mutações nos genes SCN5A e SCN10A9,10; anormalidades microscópicas estruturais e fibrose cardíacas11; arritmias ventriculares de fase 2, isto é, causadas pela justaposição de miócitos com canais de sódio normais e outros com canais de sódio defeituosos, com períodos de refratariedade diferentes, permitindo a geração de circuitos de reentrada locais12; presença de febre13, uso de cocaína ou outras drogas psicotrópicas14.
Do ponto de vista clínico, mais de 30% dos pacientes apresentam, como manifestação inicial, morte súbita de origem cardíaca, que ocorre mais comumente à noite. Podem ocorrer também síncope, palpitações (devido maior risco de fibrilação atrial15), episódios de respiração agônica durante o sono. O eletrocardiograma sempre apresenta pseudo-bloqueio de ramo direito (sem as alterações típicas do bloqueio de ramo direito em V6) e elevação do segmento ST > 2 mm em mais de uma das derivações V1 a V3; dois padrões de alterações no segmento ST são descritas: 1) Supradesnível do segmento ST desce com a convexidade voltada para baixo, até a linha de base, terminando em uma onda T invertida (figura 3); 2) O traçado entre o segmento ST e a onda T adquire um formato peculiar (“saddleback” em inglês), em que o supradesnivelamento do segmento ST desce até pelo menos 0,5 mm acima da linha de base e então ascende novamente, terminando em onda T positiva ou bifásica (figura 4). Alguns pacientes apresentam prolongamento do intervalo QT4. Já foi demonstrado que as alterações no ECG podem ser transitórias, e são induzíveis por fármacos antiarrítmicos classe ia – bloqueadores de canais de sódio – como flecainida e procainamida, beta-bloqueadores, antidepressivos tricíclicos ou tetracíclicos, lítio, anestésicos locais, alterações eletrolíticas.
Figura 3. ECG com alterações típicas de SB, com morfologia do segmento ST tipo 1. Figura retirada do site:
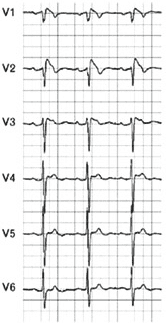
Figura 4. ECG com alterações típicas de SB, com morfologia do segmento ST tipo 2. Figura retirada do site:
O diagnóstico de SB pode ser dado baseado na presença do ECG típico e manifestações clínicas, ou seja, fibrilação ventricular documentada, taquicardia ventricular polimórfica, história familiar de morte súbita cardíaca antes dos 45 anos de idade, história familiar de alterações eletrocardiográficas compatíveis, taquicardia ventricular induzida durante estudo eletrofisiológico, síncope presumidamente causada por taquiarritmia, ou respiração noturna agônica. O diagnóstico diferencial inclui displasia arritmogênica do ventrículo direito, repolarização precoce, pericardite aguda, isquemia miocárdica, hipotermia, síndrome do QT longo, taquicardia ventricular polimórfica catecolaminérgica, síndrome do QT curto, commotio cortis4. Exames especiais como estudo eletrofisiológico, teste provocativo com antiarrítmicos classe ia, teste genético (disponível para mutações SCN5A e SCN10A) são empregados em pacientes assintomáticos com história familiar positiva ou em casos duvidosos. O teste genético é limitado pois muitos pacientes não exibem mutações nos genes estudados, e a presença da mutação genética sem clínica e com exames normais é comum.
Entre os fatores de mau prognóstico, figuram a ocorrência prévia de taquicardia ventricular com síncope ou morte súbita abortada, ocorrência de fibrilação atrial, sexo masculino, história familiar de morte súbita de origem cardíaca16.
O tratamento deve ser adequado a cada situação clínica17: pacientes com SB e morte súbita abortada ou que tiveram taquicardia ventricular espontânea e sustentada, bem como pacientes com pB e que tiveram síncope presumidamente por taquicardia ventricular devem ser submetidos ao implante de cardiodesfibrilador implantável (CDI), reservando-se o uso de fármacos (amiodarona, quinidina) para pacientes que recusam o procedimento ou com baixa expectativa de vida. Não há benefício em implantar CDI em pacientes assintomáticos com SB ou pB, e pacientes com SB, sem eventos cardíacos prévios, que apresentam taquicardia ventricular durante estudo eletrofisiológico, podem eventualmente receber CDI. Familiares de primeiro grau do paciente devem ser triados com anamnese cuidadosa e ECG em repouso18.
O paciente mostrado nesse relato possui a história clínica clássica da SB: adulto jovem, sexo masculino, usuário de entorpecentes, apresentou episódio de morte súbita de origem cardíaca abortada não relacionada com esforço físico, e apresentava vários ECG em repousos compatíveis com esta síndrome. Como já houvesse apresentado evento cardíaco, foi corretamente submetido ao implante de CDI.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Siscovick DS. Challenges in cardiac arrest research: data collection to assess outcomes. Ann Emerg Med 1993; 22:92
2. Demirovic J, Myerburg RJ. Epidemiology of sudden coronary death: an overview. Prog Cardiovasc Dis 1994; 37:39
3. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 1992; 20:1391
4. Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111:659
5. Matsuo K, Akahoshi M, Nakashima E, et al. Clinical characteristics of subjects with the Brugadatype electrocardiogram: J Cardiovasc Electrophysiol 2004; 15:653
6. Gehi AK, Duong TD, Metz LD, et al. Risk stratification of individuals with the Brugada electrocardiogram: a meta-analysis. J Cardiovasc Electrophysiol 2006; 17:577
7.Benito B, Sarkozy A, Mont L, et al. Gender differences in clinical manifestations of Brugada syndrome. J Am Coll Cardiol 2008; 52:1567
8. Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation 2002; 105:1342.
9. Priori SG, Napolitano C, Gasparini M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation 2000; 102:2509
10. Hu D, Barajas-Martínez H, Pfeiffer R, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol 2014; 64:66.
11. Frustaci A, Priori SG, Pieroni M, et al. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005; 112:3680
12. Haïssaguerre M, Extramiana F, Hocini M, et al. Mapping and ablation of ventricular fibrillation associated with long QT and Brugada syndromes. Circulation 2003; 108:925
13. Amin AS, Meregalli PG, Bardai A, et al. Fever increases the risk for cardiac arrest in the Brugada syndrome. Ann Intern Med 2008; 149:216
14. Littmann L, Monroe MH, Svenson RH. Brugada-type electrocardiographic pattern induced by cocaine. Mayo Clin Proc 2000; 75:845
15. Bordachar P, Reuter S, Garrigue S, et al. Incidence, clinical implications and prognosis of atrial arrhythmias in Brugada syndrome. Eur Heart J 2004; 25:879
16. Probst V, Veltmann C, Eckardt L, et al. Longterm prognosis of patients diagnosed with Brugada syndrome: Results from the FINGER Brugada Syndrome Registry. Circulation 2010; 121:635
17. Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013; 10:1932
18. Epstein AE, DiMarco JP, Ellenbogen KA, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2013
[1] Médica especialista em Clínica Médica pelo HUGV (Hospital Universitário Getúlio Vargas)
[2] Médica especialista em Clínica Médica e em Cardiologia pelo HUGV
[3] Médica especialista em Clínica Médica pelo HUGV
[4] Médica especialista em Cardiologia e em Ecocardiografia pelo HUGV
[5] Médico especialista em Medicina Intensiva pela AMIB (Associação de Medicina Intensiva Brasileira) e em Neurologia pelo HUGV