REVISÃO BIBLIOMÉTRICA
ROSA, Daniel Trentini Farias [1], BATISTA, Matheus Couto [2], ARAÚJO, Mayle Gomes Ferreira [3], SALVÁ, Marília de Andrade [4], OLIVEIRA, Michelle Jacintha Cavalcante [5]
ROSA, Daniel Trentini Farias. Amiloidose sistêmica primária com envolvimento renal e cutâneo: relato de caso e revisão de literatura. Revista Científica Multidisciplinar Núcleo do Conhecimento. Ano 03, Ed. 12, Vol. 06, pp. 77-85 Dezembro de 2018. ISSN:2448-0959
RESUMO
CONTEXTO: A amiloidose é uma doença rara, com incidência de 9 casos/ano por milhão de pessoas, que ocorre devido ao depósito tissular, localizado ou sistêmico, de substâncias de natureza proteica. Sua origem pode ser genética (amiloidose hereditária), primária (AL – Amiloidose Light Chain) ou secundária (AA – familiar ou não familiar). O objetivo deste estudo é relatar um caso de amiloidose primária com acometimento renal diagnosticado no Hospital Universitário Professor Alberto Antunes (HUPAA), enfatizando as dificuldades do diagnóstico e apresentando uma revisão de literatura. RELATO DO CASO: Sexo feminino, 58 anos, branca, obesa mórbida (grau III), hipertensa, foi admitida no Serviço de Clínica Médica do hospital com quadro de anasarca e urina espumosa há 6 meses. Realizou-se biópsia renal, que não positivou para vermelho-congo devido à ausência de glomérulos no fragmento enviado à microscopia, além da dificuldade técnica imposta pela obesidade da paciente. No entanto, os achados da imunofluorescência sugeriram glomeruloesclerose segmentar e focal (GESF). Com isso, seguiu-se a investigação, sendo realizada biópsia incisional de lesão palpebral, biópsia de gordura periumbilical (ambas coraram para vermelho-congo), além de imunofixação de proteína em urina de 24 horas, com resultado de eliminação de cadeias leve kappa, lambda e lambda livre, o que permitiu o diagnóstico de amiloidose. CONCLUSÃO: A amiloidose consiste em uma doença extremamente rara e de difícil diagnóstico. Isso ocorre principalmente em pacientes que possuem um quadro rico em comorbidades, devido a manifestações pouco específicas da doença. Trata-se, muitas vezes, de um diagnóstico de exclusão, sendo considerado quando a sintomatologia é rica, como nos casos de síndrome nefrótica, ou quando diversas outras doenças forem descartadas, o que pode contribuir com a evolução do quadro e perda do tempo correto de tratamento. O caso relatado refere-se a uma apresentação da amiloidose primária com envolvimento renal, como apresentação clínica inicial da doença, e as dificuldades do diagnóstico, em virtude do quadro extenso de comorbidades e da biópsia renal não corada pelo vermelho-congo.
Palavras – chave: Evolução, biópsia, diagnóstico, proteína.
INTRODUÇÃO
Amiloidose é uma doença rara, com incidência de 9 casos/ano por milhão de pessoas, acometendo 2 vezes mais o sexo masculino, com diagnóstico ocorrendo a partir dos 65 anos de idade1. Existem três tipos possíveis: a amiloidose primária (AL ou Amiloidose Light Chain), amiloidose secundária (AA – familiar ou não familiar), amiloidose hereditária (AF) ou amiloidose senil (SSA). Dentre as AA não familiares as poliartropatias inflamatórias respondem por 60% dos casos 3.
Caracterizada pela presença de depósitos amiloides nos tecidos acometidos, identificados histologicamente pela positividade ao corante vermelho do Congo e birrefringência quando vistos sob luz polarizada. A amiloidose pode ser sistêmica ou localizada. Os órgãos mais acometidos na amiloidose primária são os rins, o coração e o fígado.2
Perda de peso, parestesias, dispneia e fadiga são os sintomas mais associados a amiloidose e são comuns a todas as formas sistêmicas. No entanto, essas queixas são inespecíficas. A púrpura amilóide está presente em cerca de 1 em 6 pacientes com AL, sendo tipicamente periorbital. O acometimento renal típico cursa com síndrome nefrótica, sendo representado pela tétrade edema, hiperlipidemia, albuminúria e hipoalbuminemia.
Uma vez suspeitado, o diagnóstico deve ser confirmado através de biópsia do órgão afetado, com avaliação histopatológica pela técnica de coloração vermelho do Congo, sendo considerado o padrão ouro. Deve-se solicitar também a pesquisa de proteína monoclonal sérica ou urinária ou free light chain.4
Tanto a biópsia de tecido subcutâneo como da medula óssea são recomendadas e demonstrarão depósitos de amiloide em 85% dos pacientes. Outros tecidos que podem ser facilmente biopsiados incluem lábio, reto, artéria temporal ou pele. Se esses estudos forem negativos, a biópsia de um órgão afetado (coração, fígado, rim ou nervo) deve ser realizada.
RELATO DE CASO
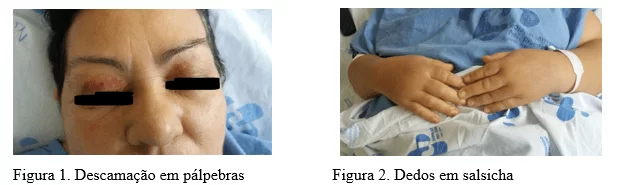
Mulher, 58 anos, viúva, do lar, branca, natural de Maceió, foi encaminhada pela reumatologia para o serviço de Nefrologia do HUPAA com queixas de anasarca, iniciada com edema progressivo em articulações, membros inferiores e, em especial, nas pálpebras (figura 1), além de urina espumosa há 6 meses. Em virtude da sua obesidade (IMC: 41,1 – obesidade mórbida – grau III), o edema era de difícil avaliação.
Paciente com diagnóstico prévio de hipertensão arterial há 20 anos, osteoartrite e osteoporose, já acompanhada pela cardiologia e reumatologia, realizando seus respectivos tratamentos (em uso de: furosemida, 40 mg/dia, losartana 100 mg/dia, espironolactona 50 mg/dia, codeína 60 mg/dia). Paciente relatou histórico recente de internamento hospitalar em outro serviço por oito dias pelos sinais e sintomas acima. Traz à consulta exames previamente solicitados, como descrito na tabela 1.
Tabela 01- Exames Laboratoriais da 1ª consulta (19/04/2017)
| Exame | Resultado | Valor de Referência |
| Proteinúria 24h | 4882 mg/24h | < 150 mg/24h |
| EAS | Proteínas (3+/4+)
Leucócitos 1/campo Hemácias 2/campo |
Proteínas –
Leucócito 5/campo Hemácias – |
| Hemoglobina | 11 g/dL | 12-16 g/dL |
| Hematócrito | 33% | 37-49% |
| Leucócitos | 13.200/mm³ | 4.000-11.000/mm³ |
| Plaquetas | 371.000/mm³ | 180-500 x 10³/mm³ |
| Ureia | 24,01 mg/dL | 10-50 mg/dL |
| Creatinina | 0,55 mg/dL | 0,6-1,0 mg/dL |
| Potássio | 3,8 mEq/L | 3,5-5,5 mEq/L |
| Sódio | 139 mEq/L | 135-145 mEq/L |
| Colesterol Total | 274 mg/dL | < 200 mg/dL |
| Triglicerídeos | 242 mg/dL | ≤ 150 mg/dL |
| HDL | 40 mg/dL | ≥ 40 mg/dL |
| LDL | 184 mg/dL | < 100 mg/dL |
| VHS | 135 mm/h | < 30 mm/h |
| COOMBS Direto | Negativo | |
| PCR | 13,8 mg/dL | < 5 mg/dL |
| Ferritina | 237 ng/mL | 15-400 ng/mL |
| Ferro Sérico | 36 mcg/dL | 50-150 mcg/dL |
| Transferrina | 193 mg/dL | 230-390 mg/dL |
| TGO | 15 U/L | < 32 U/L |
| TGP | 15 U/L | < 31 U/L |
| Fosfatase Alcalina | 222 U/L | 35- 104 U/L |
| Gama-GT | 27 U/L | < 43 U/L |
| FAN | ANTI-SM NEGATIVO
ANTI-NUCLEARES NEGATIVOS |
|
| P-ANCA | NEGATIVO | |
| C-ANCA | NEGATIVO | |
| Eletroforese de
proteínas |
Proteína 5,2 g/dL Beta 1 0,58 g/dL
Albumina 2 g/dL Beta 2 0,4 g/dL Alfa 1 0,18 g/dL Gama 0,19 g/dL Alfa 2 0,98 g/dL Relação A/G 1,15 |
|
| Sorologias | ANTI-HIV 1 e 2: NÃO REAGENTE
ANTI-HCV: NÃO REAGENTE ANTI-HBC TOTAL: NÃO REAGENTE ANTI-HBS: 0,56 HBSAG: NÃO REAGENTE VDRL: NÃO REAGENTE |
|
| C3 | 1,82 g/dL | 0,83 – 1,93 g/dL |
Ao exame físico, paciente apresentava-se ao leito, em bom estado geral, lúcida e orientada em tempo e espaço, eupneica, cooperativa, hidratada, hipocorada (+/4+), acianótica, anictérica, hemodinamicamente estável e em anasarca. À ausculta cardíaca, apresentou ritmo cardíaco regular em dois tempos, com bulhas hipofonéticas. Nas extremidades, apresentava edema (4+/4+) em membros inferiores e mãos com dedos em salsicha. Em região orbital, apresentava descamação em pálpebras bilateralmente. Foi indicada internação, prescrito diurético para diminuição do edema e compensação clínica das comorbidades, e programada a biópsia renal para o rastreio da síndrome nefrótica.
Na internação, questionavam-se as seguintes etiologias: colagenose, amiloidose e artrite erosiva. Na enfermaria da Clínica Médica, paciente continuava com quadro de anasarca, apresentava lesões equimóticas < 1 cm em pálpebras bilaterais (figura 1) e em região suprapúbica, além de eritema e descamação em região inguinal. Nos membros inferiores, havia presença de pápulas hipocrômicas, sem sinais flogísticos, não pruriginosas, além de intenso edema (4+/4+), com cacifo positivo. Em membros superiores, o edema era de 3+/4+, cacifo negativo, com dedos em salsicha (figura 2).
Para investigar a etiologia da síndrome nefrótica e a possibilidade de amiloidose, foi realizada uma biópsia renal, uma semana depois do internamento, com o seguinte resultado: “Quadro não é totalmente conclusivo devido à ausência de glomérulos no fragmento enviado para a microscopia óptica. No entanto, os achados da imunofluorescência sugerem glomeruloesclerose segmentar e focal.”. Além disso, foram realizados outros exames de imagem:
| Ultrassom de abdome total | Sinais de esteatose hepática leve |
| Ultrassom dos ombros | Tendinopatia do músculo supraespinhal e bursite |
| Tomografia Computadorizada do tórax e abdome | Edema difuso da tela subcutânea, imagem ovalada com centro hipodenso na região axilar direita (linfonodomegalia necrótica?), raros e diminutos nódulos pulmonares não calcificados, esparsos e bilaterais, além de pequena hérnia gástrica hiatal. |
| Ecocardiograma Transtorácico | Insuficiência mitral leve. FEVE 66%. Pericárdio sem alterações. Sem evidências de hipertensão pulmonar. |
Como terapia medicamentosa para melhora da insônia e das dores articulares, foi iniciada amitriptilina, e por indicação da nefrologista, aumentou-se a dose da furosemida gradativamente, mantendo-se os medicamentos anti-hipertensivos previamente prescritos devido à anasarca e controle da PA. Além disso, a corticoterapia com prednisona na dose de 1mg/kg foi iniciada para tratamento da GESF. Cerca de 20 dias após internação, a investigação seguiu-se com a realização de biópsia da pele em lesão palpebral, além da biópsia de gordura periumbilical, as quais tiveram resultado compatível com amiloidose. Após 30 dias de internação, paciente em quadro de anasarca melhorada (peso = 92,1 kg, IMC = 38,83), recebeu alta para seguimento ambulatorial com as especialidades de nefrologia, dermatologia e reumatologia.
No seguimento com a nefrologia, após laudo da biópsia que confirmou amiloidose, iniciou-se imediatamente o desmame da prednisona, que estava sendo utilizada na dose máxima (1 mg/kg – máximo de 80 mg), a fim de tratar a síndrome nefrótica. Mesmo com o desmame, a paciente evoluiu com necrose da cabeça do fêmur direto, como complicação da corticoterapia.
Após biópsia confirmatória, agendou-se uma consulta no ambulatório de Hematologia, onde foi solicitada a eletroforese de proteínas séricas e urinárias, imunofixação sérica e urinária, além da cadeia leve plasmática. Confirmou-se o resultado de cadeias leves kappa, lambda e lambda livre na imunofixação urinária.
Como tratamento, tentou-se a realização de um transplante autólogo de medula óssea. Logo, foi encaminhada para Pernambuco, pois o serviço não é disponibilizado em Alagoas. Entretanto, o transplante foi contra-indicado devido à descompensação clínica da paciente naquele momento, que apresentava necrose avascular de cabeça do fêmur direito, trombose venosa profunda e erisipela. Atualmente, está em tratamento quimioterápico com bortezomide para esquema Cybord, além do uso de clexane em virtude das complicações adquiridas.
DISCUSSÃO
A amiloidoseé um termoabrangente para um grupo heterogêneo de doenças associadas com a deposição de proteínas anormais em tecidos e órgãos. Existem pelo menos 25 proteínas diferentes associadas a amiloidose e o tipo mais comum de amiloidose e a mais associada a um mau prognóstico é a amiloidose primária, também denominada de amiloidose de cadeias leves (AL) ou a associada a mieloma múltiplo.(5)
A amiloidose AL é causada por cadeias leves (CL) de imunoglobulina anormais produzidas por plasmócitos clonais na medula óssea. As cadeias leves anormais posteriormente sofrem dobramento incorreto, agregam-se e depositam-se nos tecidos como fibrilas amiloides que se acumulam no espaço extracelular de órgãos e tecidos. Desta forma o funcionamento normal do corpo fica comprometido e os problemas sistêmicos começam a surgir.(7,8)
Os principais locais de deposição de amiloide e com repercussões clínicas mais significativas são: rim (74%), o coração (60%), o fígado (27%) e o sistema nervoso periférico (22%) e autonómico (18%). O envolvimento dos rins, inúmeras vezes apresenta-se apenas como proteinúria assintomática ou síndrome nefrótica, assim como apresentado pela paciente deste caso.(5,6)
A síndrome nefrótica é uma das manifestações iniciais mais presentes na amiloidose primária, sendo composta pelas seguintes características clínicas e laboratoriais: proteinúria maior 3,5 g na urina de 24 h (inicialmente observada neste caso, apresentando-se como “urina espumosa”), edema ou anasarca (também observado neste caso), além de hipoalbuminemia e hiperlipidemia.(2)
Outras manifestações clínicas muito sugestivas de amiloidose AL é a macroglossia, que ocorre em apenas 10% dos casos, e as manifestações mucocutâneas presente em 30-40% dos casos, sendo a púrpura a lesão mais frequente. Uma lesão típica é a púrpura periorbitária (raccoon eyes) precipitada pela manobra de Valsalva ou por pequenos traumatismos, exibida pela paciente deste relato.(2,6)
Diante dos fatores clínicos e laboratoriais e suspeitando-se então de amiloidose, o próximo passo é a confirmação por meio da biópsia do órgão afetado e a avaliação histopatológica usando a técnica do vermelho Congo (Técnica: a substância amilóide tem uma coloração vermelho/castanho observada à luz do dia, mas o diagnóstico é confirmado pela birrefringência verde-maçã observada sob um microscópio óptico polarizador).(2,3)
Neste relato o rim era o órgão mais afetado, realizou-se então a biópsia renal com resultado não conclusivo, devido à ausência de glomérulos no fragmento biopsiado e enviado a microscopia óptica. Entretanto, o fragmento da imunofluorescência sugeria GESF. Posteriormente, fez-se biópsia de outros locais, como a pele lesionada na região palpebral e da gordura periumbilical, ambas compatíveis com amiloidose.
A literatura descreve, que após confirmar a amiloidose por meio da biópsia é preciso diferenciar o tipo de amiloidose. Para isso realiza-se então um teste de imuno-histoquímica utilizando anticorpos monoclonais para o tipo específico de cadeias leves (kappa e lambda). A presença de cadeias leves monoclonais na urina e no soro é benéfica, mas nem sempre é suficiente para diagnosticar doenças sistêmicas como AL. Portanto, todos os pacientes devem realizar a eletroforese sérica (71% de sensibilidade) e urinária (84% de sensibilidade), com o intuito de revelar cadeias leves monoclonais.2,8
A amiloidose de cadeias leves AL é forma mais diagnosticada da doença e sem o tratamento, a taxa de sobrevivência média é de cerca de 12 a 18 meses. A quimioterapia é fundamental para o tratamento da amiloidose primária, buscando interromper o crescimento de células plasmáticas que produzem proteínas de cadeias leves anormais. Durante muitos anos as terapias com Melfalan ou Ciclofosfamida foram o tratamento de escolha. Atualmente, a medicação utilizada preferencialmente é o Bortezomib.7
No caso descrito, a equipe médica solicitou o transplante autólogo de células tronco, entretanto o hospital responsável pelo tratamento recusou devido a intercorrência de trombose venosa profunda (TVP) e erisipela da paciente. Realizou-se então a terapia medicamentosa de preferência, como descrito acima: o Bortezomib, um quimioterápico inibidor dos proteassomas.
CONCLUSÕES
Certamente a amiloidose é um quadro clínico raro, frequentemente negligenciado e de diagnóstico difícil, por ser um diagnóstico de exclusão. Isso ocorre por conta das manifestações clínicas iniciais inespecíficas da doença, fazendo com que a hipótese de amiloidose seja apontada apenas quando o paciente manifesta a insuficiência de algum órgão. Este caso consistiu de uma amiloidose primária ou de cadeias leves (AL) com o envolvimento inicial dos rins como apresentação mais precoce da doença e enfatizou a importância da biópsia seguida da coloração com o vermelho Congo para o diagnóstico da amiloidose.
E em conjunto, este caso reforça aos profissionais médicos, que mesmo diante de uma biópsia renal inicialmente inconclusiva para a amiloidose, deve-se persistir com a biópsia de outros sítios como a gordura periumbilical. Além de demonstrar a importância do diagnóstico e tratamento precoces, de modo a contribuir para a estabilização da doença e melhorar o cuidado e o atendimento destes pacientes.
REFERÊNCIAS
- AMILOIDOSIS SUPPORT GROUPS. Conscientização Sobre Amiloidose. 2013. Disponível em: <http://amyloidosissupport.org/>. Acesso em: 20 set. 2018.
- DE ASÚA, Diego Real et al. Systemic AA amyloidosis: epidemiology, diagnosis, and management. Clinical epidemiology, v. 6, p. 369, 2014.
- GOREVIC, Peter D et al .Overview of amyloidosis. 2018 Jul 27 In: UpToDate [Internet]. WoltersKluwer Health, 1992 – . Disponível em: https://www.uptodate.com/contents/overview-of-amyloidosis. Acesso em: 20 set. 2018.
- HALLACK NETO, Abrahão Elias et al. Primary systemic AL amyloidosis: case report and associated considerations. Hu Revista, Ufjf, Juiz de Fora, v. 4, n. 34, p.281-285, dez. 2008.
- KASPER, DL. et al. Harrison Medicina Interna. Rio de Janeiro: McGrawHill, 18ª edição, v.2,. 2013.
- LESTRE et al, Púrpura – manifestação de amiloidose sistêmica primária, Acta Med Port. 2009; 22(3):307-312.
- MELLO, Ramon Andrade Bezerra de et al. Renal failure due to primary amyloidosis: a case report and literature review. Sao Paulo Medical Journal, v. 129, n. 3, p. 176-180, 2011.
- MONTEIRO, Natalia Fernandes; DIZ, Mary Carla Estevez. Difficulties in the diagnosis of primary amyloidosis: case report. Revista Médica de Minas Gerais, [s.l.], v. 25, n. 2, p.280-286,mar.2015.GN1 Genesis Network. http://dx.doi.org/10.5935/2238-3182.20150048.
- MORIE A. Gertz, BMJ Best Practice. Amyloidosis. 2018, https://bestpractice.bmj.com
- SANCHORAWALA, Vaishali. Light-chain (AL) amyloidosis: diagnosis and treatment. Clinical Journal of the American Society of Nephrology, v. 1, n. 6, p. 1331-1341, 2006.
- RIELLA, Miguel C. Princípios de Nefrologia e Distúrbios Hidroeletrolíticos. Rio de Janeiro, Editora Guanabara Koogan Ltda, 5a Edição, 2010
[1] Graduando em Medicina pela Universidade Federal de Alagoas.
[2] Graduando em Medicina pela Universidade Federal de Alagoas.
[3] Graduando em Medicina pela Universidade Federal de Alagoas.
[4] Residente em Clínica Médica pelo Hospital Universitário Professor Alberto Antunes.
[5] Médica Docente pela Universidade Federal de Alagoas/HUPAA.
Enviado: Novembro, 2018
Aprovado: Dezembro, 2018